SYSTEMS 2 (L160) A Randomised Phase II trial of standard versus dose escalated radiotherapy in the treatment of pain in malignant pleural mesothelioma
|
|
|
- Georgiana Fletcher
- 5 years ago
- Views:
Transcription



1 SYSTEMS 2 (L160) A Randomised Phase II trial of standard versus dose escalated radiotherapy in the treatment of pain in malignant pleural mesothelioma ISRCTN No: ISRCTN Protocol No: SYSTEMS Sponsor Ref: GN13ON388 Sponsor/Co Sponsor: NHS Greater Glasgow & Clyde/ University of Glasgow Chief Investigator: Professor Anthony Chalmers Co ordinating CTU: CRUK Clinical Trials Unit, Glasgow INITIATION SLIDES (VERSION 1.0, 14 July 2016)
2 Study Details **Please note this presentation has been prepared as part of your site initiation. These slides are a compliment to the protocol. All site staff must have read and understood the protocol, and the study requirements prior to signing off the initiation acknowledgement sheet.** Study will be conducted according to ICH GCP guidelines E6 Study conducted in accordance with the EU Directive 2001/20/EC & amendments Study carried out in accordance with the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research involving Human Subject 1964 (as amended) Study carried out in accordance to Research Governance Framework
3 Study Organisation The study is being co ordinated via the Cancer Research UK Clinical Trials Unit, Glasgow Sponsor of the study is Greater Glasgow Health Board (GGHB) and University of Glasgow (GU) Chief Investigator is Professor Anthony Chalmers Study is being funded by educational grants from The June Hancock Mesothelioma Research Fund and The Beatson Cancer Charity
4 Study Team Chief Investigator: Professor Anthony Chalmers Clinical Research Fellow: Dr Miranda Ashton Project Manager: Pharmacovigilance: Sponsor Contact: Laura Alexander Lindsey Connery Paul Dearie Clinical Trial Co ordinator: Ann Peek Clinical Trial Monitor: Calum Innes
5 Study Design Design: A multicentre phase II randomised study comparing two schedules of hypofractionated radiotherapy: Dose escalated, 36 Gy in 6# over two weeks (treatment arm) and 20 Gy in 5# over one week (standard arm) Sample Size: The sample size for this study is 112 patients with a histological or multidisciplinary team (MDT) diagnosis of MPM for whom radiotherapy is indicated for pain control Patients will be recruited from 8 10 UK centres and will be identified either at MDT meetings or by clinical oncologists to whom patients have been referred for radiotherapy
6 Study Objectives/Endpoints Objectives: Primary: Establish whether dose escalated, hypo fractionated radiotherapy (36 Gy in 6#) increases the proportion of MPM patients experiencing a clinically significant improvement in pain at 5 weeks compared with standard radiotherapy (20 Gy in 5#) Secondary: Determine the relative effects of dose escalated and standard radiotherapy on: Acute toxicity at end of radiotherapy and weeks 5 and 9 Pain response at weeks 5 and 9 (BPI) Radiological response at week 9, measured by CT scan reported to modified RECIST Overall survival Quality of life at weeks 5 and 9 (EORTC QLQ C30, LC13) Exploratory: Change in strong opioid use between baseline and study endpoint Health related quality of life (HRqol) at week 9, measured by EQ5D and ICECAP SCM Potential predictive biomarkers and response biomarkers including plasma fibulin (optional) Histopathological and immunohistochemical predictive biomarkers (optional)
7 Modified RECIST Reporting Requirements All radiological investigations must be reported as per protocol / modified RECIST Source documentation of this must be available for review if the original report has had to be supplemented to bring it in line with protocol requirements CRUK CTU, Glasgow have produced a worksheet to assist with the documentation of study specific reporting and will make this available to any participating site upon request to the study monitor
8 Eligibility Criteria Inclusion Criteria Histological and/or MDT diagnosis of MPM Performance status 0 2 (ECOG) Predicted life expectancy of >12 weeks CT scan within 8 weeks of starting radiotherapy Worst Pain 4/10 (0 10 Numerical Rating Scale) Ability to provide written informed consent prior to participating in the trial and prior to any trial related procedures being performed Willingness to comply with scheduled visits, treatment plans, laboratory tests and other study procedures Patients must have a radiotherapy plan compatible with the treatment arm (30 36 Gy in 5 6 fractions) Exclusion Criteria Patients who have received anti cancer therapy within the 4 weeks prior to study entry that is likely to alter pain at the index site during the duration of the study. Patients who are planned to have further anti cancer therapy within 6 weeks post radiotherapy treatment Psychotic disorders or cognitive impairment Co existing lung tumours at the time of study entry **Please refer to section 3 of the study protocol for full details of the eligibility criteria for the study (Brief details of the key selection criteria only is detailed on this slide)** Please note there will be no exception to the eligibility requirements at the time of registration/randomisation. Queries in relation to the eligibility criteria should be addressed via contact with the CTU prior to registration/randomisation. Patients are eligible for the trial if all the inclusion criteria are met and none of the exclusion criteria apply.
9 Pre Randomisation Requirements Radiotherapy Planning before Randomisation To avoid bias in radiotherapy treatment planning and target volume delineation, patients will first be registered on to the study; then randomised AFTER completion of radiotherapy planning but prior to receiving the first radiotherapy treatment Radiotherapy plans must be acceptable for the higher dose/fractionation regimen and can be changed to the lower dose regimen if the patient is subsequently randomised to this arm Stabilisation of Pain before Randomisation To enable the effects of the radiotherapy to be determined, patients should have their pain control optimised and their pain stabilised as much as possible before randomisation. Patients should be reviewed by the local palliative care team if necessary Stable pain will be defined as an average pain score at the index site of between 4 and 8 (inclusive, 0 10 Numerical Rating Score) for a minimum of 72 hours prior to randomisation While stable pain is recommended, it is not an absolute inclusion criterion. However, patients whose worst pain score is lower than 4 at Baseline Visit 2 will no longer be eligible for the study
10 Registration & Randomisation Process Registration Check that patient fulfils all the eligibility criteria as per trial protocol Complete Registration Form Site staff must contact the Clinical Trials Unit, Glasgow to register the patient. Registration to the trial can be done by either telephone or fax on the following numbers: Tel no: Fax no: * Monday Thursday Friday, except public holidays * Faxes received outside of office hours will be processed the next working day Each patient registered will be allocated a unique 3 digit sequential patient ID number for the trial Randomisation Check that patient still fulfils all the eligibility criteria and has radiotherapy plan compatible with dose escalated arm (as per trial protocol) Complete Randomisation Form Site staff must contact the Clinical Trials Unit, Glasgow to register the patient using details above Eligible patients will be randomised to either standard or dose escalated radiotherapy Each patient randomised will be allocated a unique sequential patient ID number for the study
11 Study Treatment Details Two schedules of hypo fractionated radiotherapy: Treatment Arm: Dose escalated, 36 Gy delivered in 6 fractions over two weeks OR Standard arm: 20 Gy delivered in 5 fractions over one week **Please refer to study protocol for detailed information on study treatments, including planning and administration of radiotherapy.**
12 Radiotherapy Dose Modifications Radiotherapy Planning For patients with large volume disease or where there is a risk of severe acute toxicity there will be the option of reducing dose to 30 Gy in 5 fractions **Please refer to Appendix 2 of the study protocol for full details of radiotherapy planning** During Radiotherapy Delays in radiotherapy of up to 1 week are permitted If patients experience acute toxicity attributable to radiotherapy during radiotherapy, treatment should be discontinued. If toxicity resolves completely within one week, patients can continue at the same dose, provided measures have been taken to prevent further severe acute toxicity (e.g. antiemetics) **Please refer to section 5.6 of the study protocol for full details of treatment modifications/dose reductions/delays (Brief details in relation to treatment modifications are provided on these slides). **

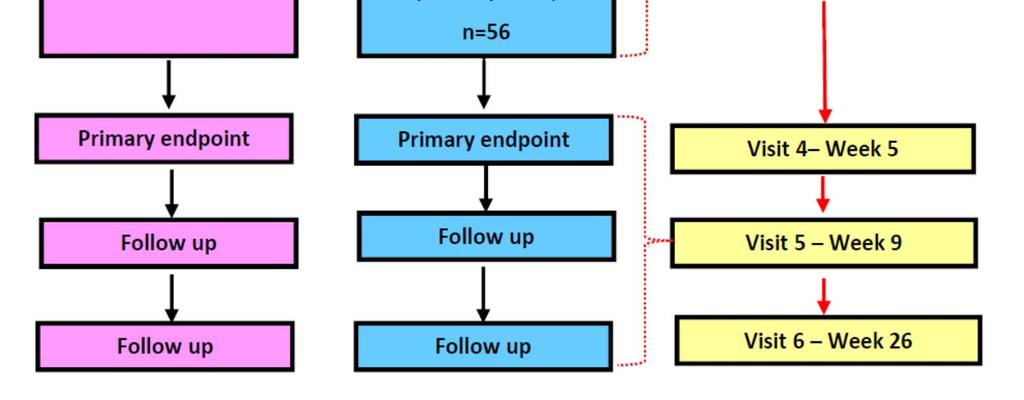
13 Visit Schedule
14 Informed Consent Process Informed consent process: Two original Consent Forms must be completed by a clinician (or deputy listed on delegation log) Two originals signed and completed by the patient Date must be prior to registration Make one photocopy Original to be filed in Investigator File Original or photocopy to be given to patient (+PIS) Photocopy to be filed in hospital notes Consent Form must not be sent to your coordinating trials office CONSENT WITHDRAWAL This is when the patient specifically asks to withdraw their consent at any point in the study. If this occurs: Document clearly in the patient notes that the patient has withdrawn consent, the level of consent withdrawal and the reason (if the patient has given any) Complete the consent withdrawal notification form Send the consent withdrawal notification form to the CRUK CTU; No further follow up should be collected on the patient from that point onwards.
15 Process for notification of protocol deviations by sites All participating sites must notify the Sponsor (via CRUK CTU) of all deviations from the protocol or GCP immediately The Sponsor requires a report on the incident(s) and a protocol deviation form will be provided during site initiation which should be used for informing of protocol deviations If site staff are unsure whether a certain occurrence constitutes a deviation from the protocol or GCP, the CRUK CTU trial team and Sponsor can be contacted immediately to discuss. The Sponsor will assess all incidents with respect to the criteria of a serious breach
16 Management of Serious Breaches The PI and site staff will be notified of any potential issues that have been identified which are considered to require escalation to the sponsor The CRUK CTU will act as the liaison between the PI and Sponsor to clarify any details or request any further information in relation to the issues Once agreed by the Sponsor that the issues are a potential serious breach or are a serious breach they will prepare the report to the Ethics Committee It is important that sites respond to requests for further information in a timely manner as serious breaches are required to be reported within 7 days of the sponsor becoming aware of issue Sponsor will onwardly report as required The Ethics Committee will respond to Sponsor and further investigation is carried out as appropriate with same staff as outlined above
17 Monitoring plan and visits Monitoring Visits/Schedule: Post initiation Monitor s check call within 10 working days of the site activation Telephone monitoring call one telephone monitoring call to be scheduled after the 1 st patient at each trial site has completed their radiotherapy course. This will be completed no later than 6 weeks post 1 st patient at site completing radiotherapy For cause monitoring visit on site visit to deal with specific issues, if required Closeout monitoring visit Telephone or on site monitoring visit at end of trial Telephone & Remote Monitoring: The time & date will be agreed with a member of the Site Study Team A pro forma covering the questions which will be covered during the telephone monitoring visit will be sent with confirmation of the agreed date Please set aside 50 to 70 minutes for this call On Site Monitoring: All patient source documentation should be made available to enable Source Document Verification by the Clinical Trial Monitor A full working day is required for on site visits & arrangements should be in place to facilitate the monitor access on the agreed date If sites are able to provide printed results/reports these must be filed in the source documents If a site is using electronic data reporting systems or electronic records &hardcopiesarenot available the clinical trial monitor must be permitted access to the system either by being issued with a temporary login or a member of staff available for the duration of the visit to facilitate electronic access to authorised reports/results All findings will be discussed at an end of visit meeting and any unresolved issues raised as Action Points Action Points will be followed up by the monitor until resolved
18 CRFs: The following Case Report Forms will be in use for this study: Registration Form Randomisation Form Pre Treatment Form End of Radiotherapy Visit Week5 and week 9 Follow up CRF Visit 6 Week 26 Follow up CRF QoL ICECAP SQM QoL EORTC QLQC30 L13 Qol EQ 5D Radiotherapy CRF Consent Withdrawal Form Pregnancy Notification Form Patient Transfer Form SAE form Data Management CRF Completion Case Report forms will be supplied electronically and these must be completed in accordance with the CRF and SAE completion guidelines issued with the CRFs. Data Escalation Process: CRUK CTU will regularly chase outstanding data from participating sites. Routine requests for outstanding data and outstanding data queries will be performed quarterly or more regularly if required for a specific study. Sites will be routinely requested to return outstanding data and data queries within 6 weeks of receiving the queries or the CRF being due for completion. Trigger reports will be run quarterly at the same point as the routine requests for data. If any site has 20%of forms overdue for more than 3 months (at least 10 forms meeting this criteria) or any forms greater than 6 months overdue the site will be contacted. A log will be kept of any sites meeting a trigger point. If a site consistently meets a trigger point an escalation process will be begin. See protocol for further information.
19 Site Set up Process CTU Glasgow REC approval Site Initiation Slides Investigator File Pharmacy File Royal Mail Safeboxes Sample Collection Kits SITE SSI (Scotland, England, Northern Ireland) or HRA Approval (England) Delegation Log R&D Approval CVs for Study Team Clinical Trial Agreement GCP Certificates for Study Team PIS, Consent, GP Letter etc on trust headed paper Initiation Process Activation of site Notification by SITE ACTIVATED
20 Confidentiality All information collected during the course of the study will be kept strictly confidential. Information will be held securely on paper and/or electronically at the CRUK CTU. The CRUK CTU will comply with all aspects of the 1998 Data Protection Act and National Health Service Guidelines for storage, transmittal and disclosure of patient information. Data on patients treated on the study will be held in study case report forms (CRFs), these files will be identified by a trial number and patient initials only. Patient identifiable data (such as full name/or initials with date of birth) should not be sent on correspondence. If you need to refer to a patient use trial and patient number. Where central monitoring of source documents by CRUK CTU (or copies of source documents) are required (e.g. scan results of blood results) the personal data of the patients must anonymised on the report e.g. black out the patient s name and any other identifiable information. Where anonymisation of documentation is required, sites are responsible for ensuring no patient identifiable data is present before sending to CRUK CTU.
21 Record Retention and Archiving Arrangements for study Archiving of the trial essential documents should be performed by both the participating trial site and Sponsor/CRUK CTU Participating sites are responsible for archiving their trial related documentation and should follow the requirements of their R&D Office in conjunction with advice from the CRUK CTU and Sponsor regarding the duration of document retention Sites should not archive their trial documentation until they have been instructed by the CRUK CTU or Sponsor that they are able to do so. Where possible, at the time of archiving, sites will be notified of the archiving retention period. If this is not confirmed at the time of archiving, sites should not destroy archived documentation until authorisation is given from the Sponsor The Sponsor and CRUK CTU will be responsible for archiving the Trial Master File (TMF) and all other essential trial documentation that is not held at participating trial sites as per their applicable SOPs
22 Pharmacovigilance Adverse Events (AEs) AEs will be recorded notified, assessed, reported, analysed and managed in accordance with the Medicines for Human Use (Clinical Trials) Regulations 2004 (as amended) and the study protocol Serious Adverse Events (SAEs) Serious Adverse Events (SAEs) must be reported to the Pharmacovigilance Office, CRUK CTU, Glasgow immediately and under no circumstances should this exceed 24 hours. SAEs are required to be reported from the start of radiotherapy treatment up to an including 30 days after the last fraction of radiotherapy. Any SAE that occurs after 30 days post radiotherapy (with no time limit) is also required to be reported if the Investigator thinks the SAE is related to protocol treatment and is medically important **For full details on Safety Reporting please see section 7 of the current study protocol** SAE References For each SAE received for a patient a SAE reference is allocated by CRUK CTU Pharmacovigilance team (for e.g. 1 for first event and 2 for patients second event ) where details of SAE are recorded on the CRF for e.g. Adverse Event table the SAE reference number allocated to the event should be recorded on the CRF. Pharmacovigilance (PV) Data Escalation Process The CRUK CTU Pharmacovigilance team will regularly chase outstanding data from participating sites in relation to SAE report forms with request for data/queries to be returned within a required timeframe. If following requests a response is not received from site staff an escalation process will begin. CRUK CTU Pharmacovigilance Team If sites have any queries they should contact the CRUK CTU Pharmacovigilance Team who will be happy to provide advice. mvls ctu pv@glasgow.ac.uk Telephone number: /7211/7953
23 Radiotherapy QA Radiotherapy Quality Assurance The radiotherapy quality assurance (RT QA) programme for the study will be designed and implemented by the National Radiotherapy Trials QA (RTTQA) Group Pre trial QA Facility questionnaire (FQ) General and trial specific questions on equipment, software and techniques to be used for the trial Dummy run QA of the outlining and planning technique for those centres where QA cannot be streamlined through a previous lung trial Dosimetry audit For IMRT delivery appropriate independent dosimetry audit evidence from centres will be required. Please contact the RTTQA group to discuss On trial QA Data collection for all patients Anonymised data, in DICOM format, will be collected by the QA team for all patients treated in the trial. This may include a brief clinical history, diagnostic imaging, full planning data including; CT images, structure set, plan and dose cube **For full details on RTTQA process please see section and Appendix 2 of the current study protocol**
24 Translational Research Requirements Blood samples Blood samples for research purposes at the Baseline visit and at weeks 5 and 9 Archival Tissue Samples Collection of surplus diagnostic tissue from the patients original biopsy (if available) for research purposes (additional biopsies or tissue samples will not be required) Consent Consent for translational research is optional. If patients do not consent to providing blood or tissue samples, it will not affect their care and they will still be eligible to participate in SYSTEMS 2 **For full details on handling, processing and shipment of these samples please see the current study Lab Manual**
25 Other Staff The Principal Investigator has overall responsibility for the conduct of the clinical trial at the trial site. BUT All staff must comply with GCP. Staff should only perform tasks delegated to them. Staff should ensure that their details are available to the Investigator. Staff should maintain appropriate confidentiality at all times
26 Contact Details for CRUK CTU, Glasgow Laura Alexander Ann Peak Project Manager Trial Coordinator Tel: Tel: Fax: Fax: E mail: laura.alexander@glasgow.ac.uk E mail: ann.peek@glasgow.ac.uk Calum Innes Trial Monitor Tel: Fax: E mail: calum.innes@glasgow.ac.uk CRUK CTU, Glasgow Cancer Research UK Clinical Trials Office Level 0, Beatson West of Scotland Cancer Centre 1053 Great Western Road Glasgow G12 0YN
STANDARD OPERATING PROCEDURE SOP 325
 STANDARD OPERATING PROCEDURE SOP 325 STUDY START UP ACTIVITIES FOR CLINICAL RESEARCH TRIALS Version 1.4 Version date 28.03.2017 Effective date 28.03.2017 Number of pages 7 Review date April 2019 Author
STANDARD OPERATING PROCEDURE SOP 325 STUDY START UP ACTIVITIES FOR CLINICAL RESEARCH TRIALS Version 1.4 Version date 28.03.2017 Effective date 28.03.2017 Number of pages 7 Review date April 2019 Author
SOP MONITORING & OVERSIGHT OF RESEARCH ACTIVITY. Contact Jess Bisset, Research Operations Manager x20227
 SOP MONITORING & OVERSIGHT OF RESEARCH ACTIVITY SETTING FOR STAFF QUERIES Trust wide All staff involved in research Contact Jess Bisset, Research Operations Manager x20227 Guidance 1. Introduction In accordance
SOP MONITORING & OVERSIGHT OF RESEARCH ACTIVITY SETTING FOR STAFF QUERIES Trust wide All staff involved in research Contact Jess Bisset, Research Operations Manager x20227 Guidance 1. Introduction In accordance
Standard Operating Procedure (SOP) Research and Development Office
 Research and Development Office") Standard Operating Procedure (SOP) Research and Development Office Title of SOP: Routine Project Audit SOP Number: 6 Version Number: 2.0 Supercedes: 1.0 Effective date: August 2013 Review date: August
Standard Operating Procedure (SOP) Research and Development Office Title of SOP: Routine Project Audit SOP Number: 6 Version Number: 2.0 Supercedes: 1.0 Effective date: August 2013 Review date: August
Standard Operating Procedure INVESTIGATOR OVERSIGHT OF RESEARCH. Chief and Principal Investigators of research sponsored and/or hosted by UHBristol
 Standard Operating Procedure INVESTIGATOR OVERSIGHT OF RESEARCH SETTING FOR STAFF ISSUE Trustwide Chief and Principal Investigators of research sponsored and/or hosted by UHBristol Oversight of research
Standard Operating Procedure INVESTIGATOR OVERSIGHT OF RESEARCH SETTING FOR STAFF ISSUE Trustwide Chief and Principal Investigators of research sponsored and/or hosted by UHBristol Oversight of research
Trial Management: Trial Master Files and Investigator Site Files
 Title: Outcome Statement: Written By: Trial Management: Trial Master Files and Investigator Site Files Staff working on research studies in NSFT will be informed about the requirements of setting up and
Title: Outcome Statement: Written By: Trial Management: Trial Master Files and Investigator Site Files Staff working on research studies in NSFT will be informed about the requirements of setting up and
I2S2 TRAINING Good Clinical Practice tips. Deirdre Thom Neonatal Nurse Coordinator
 I2S2 TRAINING Good Clinical Practice tips Deirdre Thom Neonatal Nurse Coordinator Content Principal investigator (slides 3-5) Delegation and delegation log (slides 6-7) Informed consent (slides 8-15) Data
I2S2 TRAINING Good Clinical Practice tips Deirdre Thom Neonatal Nurse Coordinator Content Principal investigator (slides 3-5) Delegation and delegation log (slides 6-7) Informed consent (slides 8-15) Data
Trial set-up, conduct and Trial Master File for HEY-sponsored CTIMPs
 R&D Department Trial set-up, conduct and Trial Master File for HEY-sponsored CTIMPs Hull And East Yorkshire Hospitals NHS Trust 2010 All Rights Reserved No part of this document may be reproduced, stored
R&D Department Trial set-up, conduct and Trial Master File for HEY-sponsored CTIMPs Hull And East Yorkshire Hospitals NHS Trust 2010 All Rights Reserved No part of this document may be reproduced, stored
SOP-QA-28 V2. Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head of School
 Title: Effective Date: 1-4-17 Review Date: 1-4-20 Author: Richard Cowie, QA Manager QA Approval: Richard Cowie, QA Manager Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head
Title: Effective Date: 1-4-17 Review Date: 1-4-20 Author: Richard Cowie, QA Manager QA Approval: Richard Cowie, QA Manager Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head
Research & Development. Case Report Form SOP. J H Pacynko and J Illingworth. Research, pharmacy and R&D staff
 Department Title of SOP Research & Development Case Report Form SOP SOP reference no: R&D GCP SOP 03 Authors: Current version number and date: J H Pacynko and J Illingworth Version 2, 01.02.18 Next review
Department Title of SOP Research & Development Case Report Form SOP SOP reference no: R&D GCP SOP 03 Authors: Current version number and date: J H Pacynko and J Illingworth Version 2, 01.02.18 Next review
STANDARD OPERATING PROCEDURE
 STANDARD OPERATING PROCEDURE Title Reference Number Sponsorship SOP-RES-001 Version Number 3 Issue Date 29 th Sep 2016 Effective Date 10 th Nov 2016 Review Date 10 th Nov 2018 Author(s) Reviewer(s) Teresa
STANDARD OPERATING PROCEDURE Title Reference Number Sponsorship SOP-RES-001 Version Number 3 Issue Date 29 th Sep 2016 Effective Date 10 th Nov 2016 Review Date 10 th Nov 2018 Author(s) Reviewer(s) Teresa
Hertfordshire Hospitals R&D Consortium Incorporating West Herts Hospitals NHS Trust and East & North Herts NHS Trust
 Hertfordshire Hospitals R&D Consortium Incorporating West Herts Hospitals NHS Trust and East & North Herts NHS Trust STANDARD OPERATING PROCEDURE FOR RESEARCH Definition of Responsibilities for Externally
Hertfordshire Hospitals R&D Consortium Incorporating West Herts Hospitals NHS Trust and East & North Herts NHS Trust STANDARD OPERATING PROCEDURE FOR RESEARCH Definition of Responsibilities for Externally
QUALITY ASSURANCE PROGRAM
 QUALITY ASSURANCE PROGRAM Elaine Armstrong, MS Quality Assurance Manager PURPOSE Verify accuracy of submitted data Verify compliance with protocol and regulatory requirements Provide educational support
QUALITY ASSURANCE PROGRAM Elaine Armstrong, MS Quality Assurance Manager PURPOSE Verify accuracy of submitted data Verify compliance with protocol and regulatory requirements Provide educational support
Version Number: 004 Controlled Document Sponsor: Controlled Document Lead:
 Chief Investigators and Principal Investigators in Research Policy CONTROLLED DOCUMENT CATEGORY: CLASSIFICATION: PURPOSE Controlled Document Number: Policy Governance To set out the responsibilities of
Chief Investigators and Principal Investigators in Research Policy CONTROLLED DOCUMENT CATEGORY: CLASSIFICATION: PURPOSE Controlled Document Number: Policy Governance To set out the responsibilities of
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES
 NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Site Selection and Initiation SOP number: TM-005 SOP category: Trial Management Version number: 04 Version date: 10 July
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Site Selection and Initiation SOP number: TM-005 SOP category: Trial Management Version number: 04 Version date: 10 July
Cancer Research UK Clinical Trials/Research Unit Glasgow. Quality System
 Cancer Research UK Clinical Trials/Research Unit Glasgow Quality System BEATSON WEST OF SCOTLAND CANCER CENTRE POLICY ON THE AWARENESS AND TRAINING OF STAFF MANAGING PATIENTS WITHIN THE CONTEXT OF A CLINICAL
Cancer Research UK Clinical Trials/Research Unit Glasgow Quality System BEATSON WEST OF SCOTLAND CANCER CENTRE POLICY ON THE AWARENESS AND TRAINING OF STAFF MANAGING PATIENTS WITHIN THE CONTEXT OF A CLINICAL
MHRA Findings Dissemination Joint Office Launch Jan Presented by: Carolyn Maloney UHL R&D Manager
 MHRA Findings Dissemination Joint Office Launch Jan. 2012 Presented by: Carolyn Maloney UHL R&D Manager Purpose of presentation To feed back abridged findings from March 2011 MHRA Statutory Systems Inspection
MHRA Findings Dissemination Joint Office Launch Jan. 2012 Presented by: Carolyn Maloney UHL R&D Manager Purpose of presentation To feed back abridged findings from March 2011 MHRA Statutory Systems Inspection
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES
 NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Site Selection and Initiation SOP number: TM 005 SOP category: Trial Management Version number: 03 Version date: 19 December
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Site Selection and Initiation SOP number: TM 005 SOP category: Trial Management Version number: 03 Version date: 19 December
ACTIONS/PSOP/001 Version 1.0 Page 2 of 6
 1. The purpose of the Pharmacy Site File To enable the designated trust pharmacy to fulfil its role and exercise appropriate control over all aspects of study medication handling, an accurately maintained
1. The purpose of the Pharmacy Site File To enable the designated trust pharmacy to fulfil its role and exercise appropriate control over all aspects of study medication handling, an accurately maintained
Document Title: Study Data SOP (CRFs and Source Data)
") Document Title: Study Data SOP (CRFs and Source Data) Document Number: SOP047 Staff involved in development: Job titles only Document author/owner: Directorate: Department: For use by: RM&G Manager, R&D
Document Title: Study Data SOP (CRFs and Source Data) Document Number: SOP047 Staff involved in development: Job titles only Document author/owner: Directorate: Department: For use by: RM&G Manager, R&D
STANDARD OPERATING PROCEDURE
 STANDARD OPERATING PROCEDURE Title Reference Number Urgent Safety Measures SOP-RES-022 Version Number 1 Issue Date 30 th April 2014 Effective Date 28 th May 2014 Review Date 28 th May 2016 Author(s) Reviewer(s)
STANDARD OPERATING PROCEDURE Title Reference Number Urgent Safety Measures SOP-RES-022 Version Number 1 Issue Date 30 th April 2014 Effective Date 28 th May 2014 Review Date 28 th May 2016 Author(s) Reviewer(s)
RITAZAREM CRF Completion Guidelines
 RITAZAREM CRF Completion Guidelines 10 Sept 2013 Version 1.2 Author: Michelle Lewin RITAZAREM Trial Coordinator Michelle.lewin@addenbrookes.nhs.uk Tel: +44(0) 1223 349350 Fax: +44(0) 1223 586767 Version
RITAZAREM CRF Completion Guidelines 10 Sept 2013 Version 1.2 Author: Michelle Lewin RITAZAREM Trial Coordinator Michelle.lewin@addenbrookes.nhs.uk Tel: +44(0) 1223 349350 Fax: +44(0) 1223 586767 Version
Investigator Site File Standard Operating Procedure (SOP)
") Investigator Site File Standard Operating Procedure (SOP) DOCUMENT CONTROL: Version: 1 Ratified by: Quality and Safety Sub Committee Date ratified: 30 January 2017 Name of originator/author: Research Nurse
Investigator Site File Standard Operating Procedure (SOP) DOCUMENT CONTROL: Version: 1 Ratified by: Quality and Safety Sub Committee Date ratified: 30 January 2017 Name of originator/author: Research Nurse
Unofficial copy not valid
 Page 2 (9) CONTENTS 1. PURPOSE... 3 2. DEFINITIONS... 3 3. RESPONSIBILITY... 3 4. INVESTIGATOR SELECTION... 3 4.1 Identification of Investigator s... 3 4.2 Initial Contacts... 4 4.3 Distribution of Pre-Study
Page 2 (9) CONTENTS 1. PURPOSE... 3 2. DEFINITIONS... 3 3. RESPONSIBILITY... 3 4. INVESTIGATOR SELECTION... 3 4.1 Identification of Investigator s... 3 4.2 Initial Contacts... 4 4.3 Distribution of Pre-Study
SOP16: Standard Operating Procedure for Establishing Sites and Centres - Site Setup
 SOP16: Standard Operating Procedure for Establishing Sites and Centres - Site Setup Authorship Team: Leanne Quinn for Joint SOP Group on Trial Processes (viz Ian Russell, Anne Seagrove, Jemma Hughes, Yvette
SOP16: Standard Operating Procedure for Establishing Sites and Centres - Site Setup Authorship Team: Leanne Quinn for Joint SOP Group on Trial Processes (viz Ian Russell, Anne Seagrove, Jemma Hughes, Yvette
Informed Consent SOP Number: 25 Version Number: 6.0 Effective Date: 1 st September 2017 Review Date: 1 st September 2019
 Standard Operating Procedures (SOP) for: Informed Consent SOP Number: 25 Version Number: 6.0 Effective Date: 1 st September 2017 Review Date: 1 st September 2019 Author: Reviewer: Reviewer: Authorisation:
Standard Operating Procedures (SOP) for: Informed Consent SOP Number: 25 Version Number: 6.0 Effective Date: 1 st September 2017 Review Date: 1 st September 2019 Author: Reviewer: Reviewer: Authorisation:
Site Closedown Checklist for UoL Sponsored CTIMP Studies
 Site Closedown Checklist for UoL Sponsored CTIMP Studies Site Information Site: Study Title: UoL study number: Centre name: Investigator: Date of Visit: Date of Report Date Responses due by: List of site
Site Closedown Checklist for UoL Sponsored CTIMP Studies Site Information Site: Study Title: UoL study number: Centre name: Investigator: Date of Visit: Date of Report Date Responses due by: List of site
Sponsor Responsibilities. Roles and Responsibilities. EU Directives. UK Law
 EU Directives Pharmacovigilance Legislation, SOPs and Reporting Louise Boldy, Governance & Safety Manager David Martin, Pharmacovigilance Monitor EU Legislation 2001/20/EC 2005/28/EC EudraLex Vol 10 UK
EU Directives Pharmacovigilance Legislation, SOPs and Reporting Louise Boldy, Governance & Safety Manager David Martin, Pharmacovigilance Monitor EU Legislation 2001/20/EC 2005/28/EC EudraLex Vol 10 UK
Quality Assurance in Clinical Research at RM/ICR. GCP Compliance Team, Clinical R&D
 Quality Assurance in Clinical Research at RM/ICR GCP Compliance Team, Clinical R&D Slide 1 of 13 What is Quality Assurance? The maintenance of a desired level of quality in a service or product, especially
Quality Assurance in Clinical Research at RM/ICR GCP Compliance Team, Clinical R&D Slide 1 of 13 What is Quality Assurance? The maintenance of a desired level of quality in a service or product, especially
Keele Clinical Trials Unit
 Keele Clinical Trials Unit Standard Operating Procedure (SOP) Summary Box Title Safety Reporting and Pharmacovigilance SOP Index Number SOP 20 Version 4.0 Approval Date 31-Jan-2017 Effective Date 14-Feb-2017
Keele Clinical Trials Unit Standard Operating Procedure (SOP) Summary Box Title Safety Reporting and Pharmacovigilance SOP Index Number SOP 20 Version 4.0 Approval Date 31-Jan-2017 Effective Date 14-Feb-2017
STANDARD OPERATING PROCEDURE SOP 715. Principles of Clinical Research Laboratory Practice
 STANDARD OPERATING PROCEDURE SOP 715 Principles of Clinical Research Laboratory Practice Version 1.2 Version date 13.11.2015 Effective date 24.04.2017 Number of pages 9 Review date June 2018 Author Role
STANDARD OPERATING PROCEDURE SOP 715 Principles of Clinical Research Laboratory Practice Version 1.2 Version date 13.11.2015 Effective date 24.04.2017 Number of pages 9 Review date June 2018 Author Role
STANDARD OPERATING PROCEDURE SOP 710. Good Clinical Practice AUDIT AND INSPECTION. NNUH UEA Joint Research Office. Acting Research Services Manager
 STANDARD OPERATING PROCEDURE SOP 710 Good Clinical Practice AUDIT AND INSPECTION Version 1.3 Version date 27.02.2018 Effective date 3.03.2018 Number of pages 10 Review date February 2020 Author Role Approved
STANDARD OPERATING PROCEDURE SOP 710 Good Clinical Practice AUDIT AND INSPECTION Version 1.3 Version date 27.02.2018 Effective date 3.03.2018 Number of pages 10 Review date February 2020 Author Role Approved
Study Monitoring Plan Template
 Study Monitoring Plan Template Sponsor Reference Number: Study Title: Principal Investigator: Study Centre: The Sponsor risk assessment form and the trial risk based monitoring strategy appendices 2 &
Study Monitoring Plan Template Sponsor Reference Number: Study Title: Principal Investigator: Study Centre: The Sponsor risk assessment form and the trial risk based monitoring strategy appendices 2 &
Keele Clinical Trials Unit
 Keele Clinical Trials Unit Standard Operating Procedure (SOP) Summary Box Title SOP Index Number SOP 21 Version 4.0 Approval Date Effective Date Non-Compliance: Deviations and Serious Breaches of GCP and/or
Keele Clinical Trials Unit Standard Operating Procedure (SOP) Summary Box Title SOP Index Number SOP 21 Version 4.0 Approval Date Effective Date Non-Compliance: Deviations and Serious Breaches of GCP and/or
COMPETENCY FRAMEWORK
 COMPETENCY FRAMEWORK Theresa Ledger Lead Nurse Research and Development Clinical Research Facility Sheffield C:\Documents and Settings\Robertus\My Documents\Mariann\CRF\CRF Portfolio and Competency Template_DRAFT
COMPETENCY FRAMEWORK Theresa Ledger Lead Nurse Research and Development Clinical Research Facility Sheffield C:\Documents and Settings\Robertus\My Documents\Mariann\CRF\CRF Portfolio and Competency Template_DRAFT
Monitoring Clinical Trials
 This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
Compliance with IR(ME)R in radiotherapy departments across England
R in radiotherapy departments across England") C Compliance with IR(ME)R in radiotherapy departments across England A summary of our programme of inspections during 2007 to 2009 January 2011 Introduction During 2007 to 2009, we carried out a programme
C Compliance with IR(ME)R in radiotherapy departments across England A summary of our programme of inspections during 2007 to 2009 January 2011 Introduction During 2007 to 2009, we carried out a programme
Standard Operating Procedure
 Standard Operating Procedure SOP number: SOP full title: SOP-JRO-07-004 Recording, managing and reporting Adverse Events for Clinical Trials of Investigational Medicinal Products and trials of Advanced
Standard Operating Procedure SOP number: SOP full title: SOP-JRO-07-004 Recording, managing and reporting Adverse Events for Clinical Trials of Investigational Medicinal Products and trials of Advanced
Standard Operating Procedure Research Governance
 Research and Enterprise Standard Operating Procedure Research Governance Title: Research Governance Audit SOP Reference Number: QUB-ADRE-08 Date prepared 7 August 008 Version Number: Final v -6.0 Revision
Research and Enterprise Standard Operating Procedure Research Governance Title: Research Governance Audit SOP Reference Number: QUB-ADRE-08 Date prepared 7 August 008 Version Number: Final v -6.0 Revision
Standard Operating Procedure (SOP)
") Standard Operating Procedure MANAGEMENT OF BREACHES IN RESEARCH SETTING AUDIENCE ISSUE Trustwide for research sponsored by UHBristol All research staff involved in UH Bristol sponsored research This SOP
Standard Operating Procedure MANAGEMENT OF BREACHES IN RESEARCH SETTING AUDIENCE ISSUE Trustwide for research sponsored by UHBristol All research staff involved in UH Bristol sponsored research This SOP
Research Governance Framework 2 nd Edition, Medicine for Human Use (Clinical Trial) Regulations 2004
 Regulations 2004") Title: Outcome Statement: Research Auditing and Monitoring Procedures Researchers in the Trust and research partners will be informed about the requirements and procedures involved in research audit and
Title: Outcome Statement: Research Auditing and Monitoring Procedures Researchers in the Trust and research partners will be informed about the requirements and procedures involved in research audit and
Document Title: Site Selection and Initiation for RFL Sponsored Studies Document Number: 026
 Document Title: Site Selection and Initiation for RFL Sponsored Studies Document Number: 026 Version: 1.1 Ratified by: Committee Date ratified: 03/10/2017 Name of originator/author: Directorate: Department:
Document Title: Site Selection and Initiation for RFL Sponsored Studies Document Number: 026 Version: 1.1 Ratified by: Committee Date ratified: 03/10/2017 Name of originator/author: Directorate: Department:
CLINICAL RESEARCH POLICY
 CLINICAL RESEARCH POLICY Approved by: Date of approval: Originator: Medical Director POLICY STATEMENT Good quality clinical research is important for furthering our understanding of the problems encountered
CLINICAL RESEARCH POLICY Approved by: Date of approval: Originator: Medical Director POLICY STATEMENT Good quality clinical research is important for furthering our understanding of the problems encountered
The Principal Investigator Role
 The Principal Investigator Role Jo Rodda Consultant in Old Age Psychiatry, NELFT North Thames CRN Dementia Specialty Lead What is a Principal Investigator? The person responsible for the conduct of a research
The Principal Investigator Role Jo Rodda Consultant in Old Age Psychiatry, NELFT North Thames CRN Dementia Specialty Lead What is a Principal Investigator? The person responsible for the conduct of a research
Document Title: Investigator Site File. Document Number: 019
 Document Title: Investigator Site File Document Number: 019 Version: 1.1 Ratified by: R&D Committee Date ratified: 03/10/2017 Name of originator/author: Directorate: Department: Name of responsible individual:
Document Title: Investigator Site File Document Number: 019 Version: 1.1 Ratified by: R&D Committee Date ratified: 03/10/2017 Name of originator/author: Directorate: Department: Name of responsible individual:
managing or activities.
 STANDARD OPERATING PROCEDURE Clinical Research Monitoring TITLE: Site Initiation Visit TITLE: Site Initiation Visit 1. PURPOSE SOP Number: Version: 1.0 MICHR CRM MON 002 Effective Date: 19Dec2013 1.1 This
STANDARD OPERATING PROCEDURE Clinical Research Monitoring TITLE: Site Initiation Visit TITLE: Site Initiation Visit 1. PURPOSE SOP Number: Version: 1.0 MICHR CRM MON 002 Effective Date: 19Dec2013 1.1 This
Northern Ireland Peer Review of Cancer MDTs. EVIDENCE GUIDE FOR LUNG MDTs
 Northern Ireland Peer Review of Cancer MDTs EVIDENCE GUIDE FOR LUNG MDTs CONTENTS PAGE A. Introduction... 3 B. Key questions for an MDT... 6 C. The Review of Clinical Aspects of the Service... 8 D. The
Northern Ireland Peer Review of Cancer MDTs EVIDENCE GUIDE FOR LUNG MDTs CONTENTS PAGE A. Introduction... 3 B. Key questions for an MDT... 6 C. The Review of Clinical Aspects of the Service... 8 D. The
Standard Operating Procedure (SOP) Research and Development Office
 Research and Development Office") Standard Operating Procedure (SOP) Research and Development Office Title of SOP: Delegated Responsibilities in Research Projects SOP Number: 11 Version Number: 2.0 Supercedes: 1.0 Effective date: August
Standard Operating Procedure (SOP) Research and Development Office Title of SOP: Delegated Responsibilities in Research Projects SOP Number: 11 Version Number: 2.0 Supercedes: 1.0 Effective date: August
STH Researcher. Recording of research information in patient case notes
 STANDARD OPERATING PROCEDURE STH Researcher Recording of research information in patient case notes SOP History None SOP Number A108 Created Research Department (AL) SUPERSEDED Final 1.3 Version 3.5 Date
STANDARD OPERATING PROCEDURE STH Researcher Recording of research information in patient case notes SOP History None SOP Number A108 Created Research Department (AL) SUPERSEDED Final 1.3 Version 3.5 Date
Joint Statement on the Application of Good Clinical Practice to Training for Researchers
 Joint Statement on the Application of Good Clinical Practice to Training for Researchers HRA, MHRA, Devolved Administrations for Northern Ireland, Scotland and Wales v1.1 12/10/17 Summary This joint statement
Joint Statement on the Application of Good Clinical Practice to Training for Researchers HRA, MHRA, Devolved Administrations for Northern Ireland, Scotland and Wales v1.1 12/10/17 Summary This joint statement
Standard Operating Procedure. Essential Documents: Setting Up a Trial Master File. SOP effective: 19 February 2016 Review date: 19 February 2018
 Standard Operating Procedure SOP number: SOP full title: SOP-JRO-06-003 Essential Documents: Setting Up a Trial Master File SOP effective: 19 February 2016 Review date: 19 February 2018 SOP author signature:
Standard Operating Procedure SOP number: SOP full title: SOP-JRO-06-003 Essential Documents: Setting Up a Trial Master File SOP effective: 19 February 2016 Review date: 19 February 2018 SOP author signature:
4.2. Clinical Trial Monitor (or Monitor): The person responsible for monitoring the data on behalf of the sponsor or contract research organization.
: The person responsible for monitoring the data on behalf of the sponsor or contract research organization.") SOP #: MON-101 Page: 1 of 6 1. POLICY STATEMENT: The DF/HCC understands that external sponsors are required to monitor the progress of clinical investigations and ensure appropriate research data collection
SOP #: MON-101 Page: 1 of 6 1. POLICY STATEMENT: The DF/HCC understands that external sponsors are required to monitor the progress of clinical investigations and ensure appropriate research data collection
Experiences of using routinely collected medical data in a cardiovascular safety trial?
 Experiences of using routinely collected medical data in a cardiovascular safety trial? Ian Ford Robertson Centre for Biostatistics and Glasgow Clinical Trials Unit Issues in clinical trials Study feasibility
Experiences of using routinely collected medical data in a cardiovascular safety trial? Ian Ford Robertson Centre for Biostatistics and Glasgow Clinical Trials Unit Issues in clinical trials Study feasibility
Details: Approval: Distribution & Storage: Pharmacovigilance for Researchers for UoL / LTHT Sponsored CTIMPs. Standard Operating Procedure
 Details: Author: Razwan Mahroof - QA Clinical Trials Monitor SOP Pages: 10 Version No. of replaced SOP: 1.0 Effective date of replaced SOP: 04 December 2015 Approval: Version No: of the SOP being approved.
Details: Author: Razwan Mahroof - QA Clinical Trials Monitor SOP Pages: 10 Version No. of replaced SOP: 1.0 Effective date of replaced SOP: 04 December 2015 Approval: Version No: of the SOP being approved.
Governance %%.4- r2&% Queen s University Belfast. Standard Operating Procedure Research Governance. r2.aoc7. Research and Enterprise
 Queen s University Belfast Research and Enterprise Standard Operating Procedure Research Governance Title: Delegation of Responsibilities SOP Reference QUB-ADRE-005 Date prepared 23 June 2008 Number: Version
Queen s University Belfast Research and Enterprise Standard Operating Procedure Research Governance Title: Delegation of Responsibilities SOP Reference QUB-ADRE-005 Date prepared 23 June 2008 Number: Version
Initially Submitted on 11/24/2009 Final Submission By Test6 CA on 11/24/2009 1:51 PM Approval By student13 student13 on 11/24/2009 1:52 PM Attendees
 Location: Los Angeles Research Institute: Los Angeles, CA Investigation Product/Test Article: Laftr Visit Mechanism: On-site Initially Submitted on 11/24/2009 Final Submission By Test6 CA on 11/24/2009
Location: Los Angeles Research Institute: Los Angeles, CA Investigation Product/Test Article: Laftr Visit Mechanism: On-site Initially Submitted on 11/24/2009 Final Submission By Test6 CA on 11/24/2009
STANDARD OPERATING PROCEDURE
 STANDARD OPERATING PROCEDURE Title Reference Number Adverse Event Identification, Recording and Reporting in Clinical Trials of Investigational Medicinal SOP-RES-019 Version Number 2 Issue Date 08 th Dec
STANDARD OPERATING PROCEDURE Title Reference Number Adverse Event Identification, Recording and Reporting in Clinical Trials of Investigational Medicinal SOP-RES-019 Version Number 2 Issue Date 08 th Dec
MANAGEMENT OF PROTOCOL AND GCP DEVIATIONS AND VIOLATIONS
 MANAGEMENT OF PROTOCOL AND GCP DEVIATIONS AND VIOLATIONS DOCUMENT NO.: CR010 v4.0 AUTHOR: Heather Charles ISSUE DATE: 01 September 2016 EFFECTIVE DATE: 15 September 2016 1 INTRODUCTION 1.1 The Academic
MANAGEMENT OF PROTOCOL AND GCP DEVIATIONS AND VIOLATIONS DOCUMENT NO.: CR010 v4.0 AUTHOR: Heather Charles ISSUE DATE: 01 September 2016 EFFECTIVE DATE: 15 September 2016 1 INTRODUCTION 1.1 The Academic
SOP: New Revised Reviewed Effective Date: 08 October Approved by : Supervisor/Manager Risk/Ethics Sr. Mgmt Committees Board/Councils
 Title: Assessment of Study Feasibility Manual: RI MUHC Policies and Procedures Human Research SOPs Originating Dept/ Service: RI MUHC Division of Clinical Research SOP: New Revised Reviewed Effective Date:
Title: Assessment of Study Feasibility Manual: RI MUHC Policies and Procedures Human Research SOPs Originating Dept/ Service: RI MUHC Division of Clinical Research SOP: New Revised Reviewed Effective Date:
SOP18b: Standard Operating Procedure for Preparing for External Audit and Inspection
 SOP18b: Standard Operating Procedure for Preparing for External Audit and Inspection Authorship Team: Jemma Hughes, Tina Morgan, for Joint SOP Group on Trial Processes (viz Leanne Quinn, Ian Russell, Anne
SOP18b: Standard Operating Procedure for Preparing for External Audit and Inspection Authorship Team: Jemma Hughes, Tina Morgan, for Joint SOP Group on Trial Processes (viz Leanne Quinn, Ian Russell, Anne
MEDICINES FOR HUMAN USE (CLINICAL TRIALS) REGULATIONS Memorandum of understanding between MHRA, COREC and GTAC
 REGULATIONS Memorandum of understanding between MHRA, COREC and GTAC") MEDICINES FOR HUMAN USE (CLINICAL TRIALS) REGULATIONS 2004 Memorandum of understanding between MHRA, COREC and GTAC 1. Purpose and scope 1.1 Regulation 27A of the Medicines for Human Use (Clinical Trials)
MEDICINES FOR HUMAN USE (CLINICAL TRIALS) REGULATIONS 2004 Memorandum of understanding between MHRA, COREC and GTAC 1. Purpose and scope 1.1 Regulation 27A of the Medicines for Human Use (Clinical Trials)
Reference Number: UHB 253 Version Number: 1 Date of Next Review: 22/01/2018 Previous Trust/LHB Reference Number: SR-RG-015
 Reference Number: UHB 253 Version Number: 1 Date of Next Review: 22/01/2018 Previous Trust/LHB Reference Number: SR-RG-015 Safety Reporting in CTIMPs Standard Operating Procedure Introduction and Aim The
Reference Number: UHB 253 Version Number: 1 Date of Next Review: 22/01/2018 Previous Trust/LHB Reference Number: SR-RG-015 Safety Reporting in CTIMPs Standard Operating Procedure Introduction and Aim The
VCU Clinical Research Quality Assurance Assessment
 VCU Clinical Research Quality Assurance Assessment Principal Investigator Protocol Title Protocol IRB Number Name of Person Completing Assessment Date Assessment was Completed The goal of this assessment
VCU Clinical Research Quality Assurance Assessment Principal Investigator Protocol Title Protocol IRB Number Name of Person Completing Assessment Date Assessment was Completed The goal of this assessment
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES
 NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Safety Reporting in CTIMPs and ATMPs SOP number: TM-003 SOP category: Trial Management Version number: 04 Version date:
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Safety Reporting in CTIMPs and ATMPs SOP number: TM-003 SOP category: Trial Management Version number: 04 Version date:
STANDARD OPERATING PROCEDURE SOP 205
 STANDARD OPERATING PROCEDURE SOP 205 Adverse Events: Identifying, Recording and Reporting for CTIMPs Sponsored by the Norfolk and Norwich University Hospital NHS Foundation Trust Version 2.3 Version date
STANDARD OPERATING PROCEDURE SOP 205 Adverse Events: Identifying, Recording and Reporting for CTIMPs Sponsored by the Norfolk and Norwich University Hospital NHS Foundation Trust Version 2.3 Version date
GCP INSPECTION CHECKLIST
 (This list is not all inclusive; item may be added &/or deleted as per the Study/Site/Sponsor/Lab) I. General. Name and address of the clinical trial site Tel. No. & e- mail:. Date of Inspection. Inspection
(This list is not all inclusive; item may be added &/or deleted as per the Study/Site/Sponsor/Lab) I. General. Name and address of the clinical trial site Tel. No. & e- mail:. Date of Inspection. Inspection
General Administration GA STANDARD OPERATING PROCEDURE FOR Sponsor Responsibility and Delegation of Responsibility
 General Administration GA 102.01 STANDARD OPERATING PROCEDURE FOR Sponsor Responsibility and Delegation of Responsibility Approval: Nancy Paris, MS, FACHE President and CEO (17 July 2014) (Signature and
General Administration GA 102.01 STANDARD OPERATING PROCEDURE FOR Sponsor Responsibility and Delegation of Responsibility Approval: Nancy Paris, MS, FACHE President and CEO (17 July 2014) (Signature and
STANDARD OPERATING PROCEDURE
 STANDARD OPERATING PROCEDURE Title Reference Number Corrective and Preventative Action SOP-QMS-008 Version Number 2 Issue Date 29 th Sep 2016 Effective Date 10 th Nov 2016 Review Date 10 th Nov 2018 Author(s)
STANDARD OPERATING PROCEDURE Title Reference Number Corrective and Preventative Action SOP-QMS-008 Version Number 2 Issue Date 29 th Sep 2016 Effective Date 10 th Nov 2016 Review Date 10 th Nov 2018 Author(s)
IDENTIFYING, RECORDING AND REPORTING ADVERSE EVENTS FOR CLINICAL INVESTIGATIONS OF MEDICAL DEVICES
 IDENTIFYING, RECORDING AND REPORTING ADVERSE EVENTS FOR CLINICAL INVESTIGATIONS OF MEDICAL DEVICES DOCUMENT NO.: CR012 v2.0 AUTHOR: Raymond French ISSUE DATE: 18 September 2017 EFFECTIVE DATE: 02 October
IDENTIFYING, RECORDING AND REPORTING ADVERSE EVENTS FOR CLINICAL INVESTIGATIONS OF MEDICAL DEVICES DOCUMENT NO.: CR012 v2.0 AUTHOR: Raymond French ISSUE DATE: 18 September 2017 EFFECTIVE DATE: 02 October
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES
 NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Safety Reporting in CTIMPs and ATMPs SOP number: TM 003 SOP category: Trial Management Version number: 03 Version date:
NEWCASTLE CLINICAL TRIALS UNIT STANDARD OPERATING PROCEDURES SOP details SOP title: Safety Reporting in CTIMPs and ATMPs SOP number: TM 003 SOP category: Trial Management Version number: 03 Version date:
European Medicines Agency Inspections ANNEX V TO PROCEDURE FOR CONDUCTING GCP INSPECTIONS REQUESTED BY THE EMEA: PHASE I UNITS
 European Medicines Agency Inspections London, 23 July 2008 EMEA/INS/GCP/197215/2005 Procedure no.: INS/GCP/3/V ANNEX V TO PROCEDURE FOR CONDUCTING GCP INSPECTIONS REQUESTED BY THE EMEA: PHASE I UNITS GCP
European Medicines Agency Inspections London, 23 July 2008 EMEA/INS/GCP/197215/2005 Procedure no.: INS/GCP/3/V ANNEX V TO PROCEDURE FOR CONDUCTING GCP INSPECTIONS REQUESTED BY THE EMEA: PHASE I UNITS GCP
Standard Operating Procedures (SOP) Research and Development Office
 Research and Development Office") Standard Operating Procedures (SOP) Research and Development Office Title of SOP: Principles of Data Collection and Storage SOP Number: 8 Supercedes: 1.0 Effective date: August 2013 Review date: August
Standard Operating Procedures (SOP) Research and Development Office Title of SOP: Principles of Data Collection and Storage SOP Number: 8 Supercedes: 1.0 Effective date: August 2013 Review date: August
EMA Inspection Site perspective
 EMA Inspection Site perspective Hermien Gous Wits RHI Shandukani Research Centre 27.09.2016 Cape Town Why were we inspected times? Pharmaceutical company applied for registration of the study drug in a
EMA Inspection Site perspective Hermien Gous Wits RHI Shandukani Research Centre 27.09.2016 Cape Town Why were we inspected times? Pharmaceutical company applied for registration of the study drug in a
CLOSE OUT VISIT REPORT (NO CRF TO MONITOR)
") Date: Page: 1 of 8 CLOSE OUT VISIT REPORT (NO CRF TO MONITOR) Protocol: PI Name: PI Address: Date of Visit: Monitor(s): Other Sponsor Personnel Present: Site Personnel Present at Visit (include names and
Date: Page: 1 of 8 CLOSE OUT VISIT REPORT (NO CRF TO MONITOR) Protocol: PI Name: PI Address: Date of Visit: Monitor(s): Other Sponsor Personnel Present: Site Personnel Present at Visit (include names and
Document Title: GCP Training for Research Staff. Document Number: SOP 005
 Document Title: GCP Training for Research Staff Document Number: SOP 005 Version: 2 Ratified by: Version 2, 04/10/2017 Page 1 of 13 Committee Date ratified: 26/10/2017 Name of originator/author: Directorate:
Document Title: GCP Training for Research Staff Document Number: SOP 005 Version: 2 Ratified by: Version 2, 04/10/2017 Page 1 of 13 Committee Date ratified: 26/10/2017 Name of originator/author: Directorate:
Once the feasibility assessment has been conducted the study team will be notified via (Appendix 3) of the outcome and whether the study is;
 of the outcome and whether the study is;") 1. INTRODUCTION 2. SCOPE Feasibility assessments will ascertain any operational concerns about a research study which may delay NHS Permission or cause issues with study delivery. By conducting an assessment
1. INTRODUCTION 2. SCOPE Feasibility assessments will ascertain any operational concerns about a research study which may delay NHS Permission or cause issues with study delivery. By conducting an assessment
PFF Patient Registry Protocol Version 1.0 date 21 Jan 2016
 PFF Patient Registry Protocol Version 1.0 date 21 Jan 2016 Contents SYNOPSIS...3 Background...4 Significance...4 OBJECTIVES & SPECIFIC AIMS...5 Objective...5 Specific Aims... 5 RESEARCH DESIGN AND METHODS...6
PFF Patient Registry Protocol Version 1.0 date 21 Jan 2016 Contents SYNOPSIS...3 Background...4 Significance...4 OBJECTIVES & SPECIFIC AIMS...5 Objective...5 Specific Aims... 5 RESEARCH DESIGN AND METHODS...6
Guideline for the notification of serious breaches of Regulation (EU) No 536/2014 or the clinical trial protocol
 No 536/2014 or the clinical trial protocol") 1 2 31 January 2017 EMA/430909/2016 3 4 5 Guideline for the notification of serious breaches of Regulation (EU) No 536/2014 or Draft Adopted by GCP Inspectors Working Group (GCP IWG) 30 January 2017 Adopted
1 2 31 January 2017 EMA/430909/2016 3 4 5 Guideline for the notification of serious breaches of Regulation (EU) No 536/2014 or Draft Adopted by GCP Inspectors Working Group (GCP IWG) 30 January 2017 Adopted
The GCP Perspective on Study Monitoring
 The GCP Perspective on Study Monitoring Heidi Judge, CCRP Sr. Clinical Trials Project Manager Clinical Trials Network and Institute Massachusetts General Hospital 1 Overview Monitoring Basics Who, What,
The GCP Perspective on Study Monitoring Heidi Judge, CCRP Sr. Clinical Trials Project Manager Clinical Trials Network and Institute Massachusetts General Hospital 1 Overview Monitoring Basics Who, What,
CLINICAL TRIAL COORDINATOR Clinical Trials Unit Beatson West of Scotland Cancer Centre
 CLINICAL TRIAL COORDINATOR Clinical Trials Unit Beatson West of Scotland Cancer Centre Job Reference: 0000046503G Closing Date: 7 th April 2017 Dear applicant, Thank you for your interest in this post
CLINICAL TRIAL COORDINATOR Clinical Trials Unit Beatson West of Scotland Cancer Centre Job Reference: 0000046503G Closing Date: 7 th April 2017 Dear applicant, Thank you for your interest in this post
European network of paediatric research (EnprEMA)
") 17 February 2012 EMA/77450/2012 Human Medicines Development and Evaluation Recognition criteria for self assessment The European Medicines Agency is tasked with developing a European paediatric network
17 February 2012 EMA/77450/2012 Human Medicines Development and Evaluation Recognition criteria for self assessment The European Medicines Agency is tasked with developing a European paediatric network
Good Clinical Practice: A Ground Level View
 Good Clinical Practice: A Ground Level View Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Clinical Research Administration Research Institute,
Good Clinical Practice: A Ground Level View Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Clinical Research Administration Research Institute,
This Agreement dated DD/MM/YYYY (the Effective Date ) is between
 is between") Clinical Trial Delegation of Sponsorship Responsibilities to Chief This Agreement dated DD/MM/YYYY (the Effective Date ) is between Nottingham University Hospitals NHS Trust, Derby Road, Nottingham, NG7
Clinical Trial Delegation of Sponsorship Responsibilities to Chief This Agreement dated DD/MM/YYYY (the Effective Date ) is between Nottingham University Hospitals NHS Trust, Derby Road, Nottingham, NG7
Marie-Claire Rickard, RG and GCP Manager Jimena Lovos, Quality Assurance Manager Elizabeth Clough, R&D Governance Operations Manager
 Standard Operating Procedures (SOP) for: Pharmacovigilance processing for the JRMO SOP Number: 26c Version Number: V1 Effective Date: 5/8/16 Review Date: 5/8/17 Author: Reviewer: Reviewer: Authorisation:
Standard Operating Procedures (SOP) for: Pharmacovigilance processing for the JRMO SOP Number: 26c Version Number: V1 Effective Date: 5/8/16 Review Date: 5/8/17 Author: Reviewer: Reviewer: Authorisation:
Participant Information Sheet Adults
 Participant Information Sheet Adults Prediction of Lupus TreAtment response Study (PLANS) Finding factors to help us treat lupus patients better and smarter. We would like to invite you
Participant Information Sheet Adults Prediction of Lupus TreAtment response Study (PLANS) Finding factors to help us treat lupus patients better and smarter. We would like to invite you
Standard Operating Procedure (SOP) for Reporting Serious Breaches in Clinical Research
 for Reporting Serious Breaches in Clinical Research") Standard Operating Procedure (SOP) for Reporting Serious Breaches in Clinical Research For Completion by SOP Author Reference Number PHT/RDSOP/002 Version V2.0 07 Apr 2016 Document Author(s) Document Reviewer(s)
Standard Operating Procedure (SOP) for Reporting Serious Breaches in Clinical Research For Completion by SOP Author Reference Number PHT/RDSOP/002 Version V2.0 07 Apr 2016 Document Author(s) Document Reviewer(s)
NN SS 401 NEURONEXT NETWORK STANDARD OPERATING PROCEDURE FOR SITE SELECTION AND QUALIFICATION
 NN SS 401 NEURONEXT NETWORK STANDARD OPERATING PROCEDURE FOR SITE SELECTION AND QUALIFICATION SOP: NN SS 401 Version No.: 2.0 Effective Date: 21Oct2016 SITE SELECTION AND QUALIFICATION Supercedes Document:
NN SS 401 NEURONEXT NETWORK STANDARD OPERATING PROCEDURE FOR SITE SELECTION AND QUALIFICATION SOP: NN SS 401 Version No.: 2.0 Effective Date: 21Oct2016 SITE SELECTION AND QUALIFICATION Supercedes Document:
GCP: Investigator Responsibilities. Susan Tebbs Nicola Kaganson
 GCP: Investigator Responsibilities Susan Tebbs Nicola Kaganson Investigator Responsibilities Qualifications & agreements Resources Responsibilities to the subject Ethics The protocol The IMP & randomisation
GCP: Investigator Responsibilities Susan Tebbs Nicola Kaganson Investigator Responsibilities Qualifications & agreements Resources Responsibilities to the subject Ethics The protocol The IMP & randomisation
Research & Development Quality Manual
 Title: Effective Date: 1-4-17 Review Date: 1-4-20 Author: Richard Cowie, QA Manager Version: 3 Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head of School Document History
Title: Effective Date: 1-4-17 Review Date: 1-4-20 Author: Richard Cowie, QA Manager Version: 3 Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head of School Document History
This is a full time post offered on a fixed-term basis until 31 August 2019.
 Faculty of Medicine and Health Sciences Norwich Medical School CTU Trial Assistant REF: SC3019 This is a full time post offered on a fixed-term basis until 31 August 2019. The Post A Clinical Trial Assistant
Faculty of Medicine and Health Sciences Norwich Medical School CTU Trial Assistant REF: SC3019 This is a full time post offered on a fixed-term basis until 31 August 2019. The Post A Clinical Trial Assistant
Auditing of Clinical Trials
 Version 1.2 Effective date: 3 September 2012 Author: Approved by: Claire Daffern, QA Manager Dr Sarah Duggan, CTU Manager Revision Chronology: Effective Date Version 1.2 3 Sept 2012 Version 1.1 12 May
Version 1.2 Effective date: 3 September 2012 Author: Approved by: Claire Daffern, QA Manager Dr Sarah Duggan, CTU Manager Revision Chronology: Effective Date Version 1.2 3 Sept 2012 Version 1.1 12 May
AN OVERVIEW OF CLINICAL STUDY TASKS AND ACTIVITIES
 1 AN OVERVIEW OF CLINICAL STUDY TASKS AND ACTIVITIES Key Clinical Study Tasks and Activities 2 Discussion of Key Tasks and Activities 3 Development of the Clinical Protocol and Study Materials 3 Qualification
1 AN OVERVIEW OF CLINICAL STUDY TASKS AND ACTIVITIES Key Clinical Study Tasks and Activities 2 Discussion of Key Tasks and Activities 3 Development of the Clinical Protocol and Study Materials 3 Qualification
COMMISSIONING SUPPORT PROGRAMME. Standard operating procedure
 NATIONAL INSTITUTE FOR HEALTH AND CARE EXCELLENCE COMMISSIONING SUPPORT PROGRAMME Standard operating procedure April 2018 1. Introduction The Commissioning Support Programme (CSP) at NICE supports the
NATIONAL INSTITUTE FOR HEALTH AND CARE EXCELLENCE COMMISSIONING SUPPORT PROGRAMME Standard operating procedure April 2018 1. Introduction The Commissioning Support Programme (CSP) at NICE supports the
NHS RESEARCH PASSPORT POLICY AND PROCEDURE
 LEEDS BECKETT UNIVERSITY NHS RESEARCH PASSPORT POLICY AND PROCEDURE www.leedsbeckett.ac.uk/staff 1. Introduction This policy aims to clarify the circumstances in which an NHS Honorary Research Contract
LEEDS BECKETT UNIVERSITY NHS RESEARCH PASSPORT POLICY AND PROCEDURE www.leedsbeckett.ac.uk/staff 1. Introduction This policy aims to clarify the circumstances in which an NHS Honorary Research Contract
Version Number: 003. On: September 2017 Review Date: September 2020 Distribution: Essential Reading for: Information for: Page 1 of 13
 CONTROLLED DOCUMENT Reporting Research Incidents and Breaches Policy CATEGORY: CLASSIFICATION: PURPOSE Controlled Number: Document Policy Governance To set out the framework and principles for reporting
CONTROLLED DOCUMENT Reporting Research Incidents and Breaches Policy CATEGORY: CLASSIFICATION: PURPOSE Controlled Number: Document Policy Governance To set out the framework and principles for reporting
NCCP Guidance on the Retention and Disposal of Systemic Anti-Cancer Therapy (SACT) prescriptions and compounding worksheets.
 prescriptions and compounding worksheets.") NCCP Guidance on the Retention and Disposal of Systemic Anti-Cancer Therapy (SACT) prescriptions and compounding worksheets. Version Date Amendment Approved By 1 11/01/2017 Version 1 NCCP following consultation
NCCP Guidance on the Retention and Disposal of Systemic Anti-Cancer Therapy (SACT) prescriptions and compounding worksheets. Version Date Amendment Approved By 1 11/01/2017 Version 1 NCCP following consultation
Standard Operating Procedure (SOP) Research and Development Office
 Research and Development Office") Standard Operating Procedure (SOP) Research and Development Office Title of SOP: Recording and Reporting Deviations, Violations, Potential Serious Breaches, Serious Breaches and Urgent Safety Measures
Standard Operating Procedure (SOP) Research and Development Office Title of SOP: Recording and Reporting Deviations, Violations, Potential Serious Breaches, Serious Breaches and Urgent Safety Measures
Good Documentation Practices. Human Subject Research. for
 Good Documentation Practices for Human Subject Research Bridget M. Psicihulis, RHIA, CCRC Quality Improvement Unit Coordinator Human Research Protection Program Wheaton Franciscan Healthcare (last updated
Good Documentation Practices for Human Subject Research Bridget M. Psicihulis, RHIA, CCRC Quality Improvement Unit Coordinator Human Research Protection Program Wheaton Franciscan Healthcare (last updated
Document Number: 006. Version: 1. Date ratified: Name of originator/author: Heidi Saunders, Senior Portfolio Coordinator
 including Roles and Responsibilities for the Conduct of Research Studies and Clinical Trials including CTIMPs (Clinical Trials of Investigational Medicinal Products) Document Number: 006 Version: 1 Ratified
including Roles and Responsibilities for the Conduct of Research Studies and Clinical Trials including CTIMPs (Clinical Trials of Investigational Medicinal Products) Document Number: 006 Version: 1 Ratified
Inspection report. Inspection of compliance with the Ionising Radiation (Medical Exposure) Regulations 2000:
 Regulations 2000:") Inspection report Inspection of compliance with the Ionising Radiation (Medical Exposure) Regulations 2000: University Hospitals Coventry and Warwickshire NHS Trust Date of inspection: 21 October 2008
Inspection report Inspection of compliance with the Ionising Radiation (Medical Exposure) Regulations 2000: University Hospitals Coventry and Warwickshire NHS Trust Date of inspection: 21 October 2008
Human Research Governance Review Policy
 Policy Document Title: Document ID: Document Name: Human Research Governance Review Policy PY-RSH-300304 Human Research Governance Review Policy Version Number: 2 Revision Date: Key Words 28/10/2014 10:40:00
Policy Document Title: Document ID: Document Name: Human Research Governance Review Policy PY-RSH-300304 Human Research Governance Review Policy Version Number: 2 Revision Date: Key Words 28/10/2014 10:40:00