Clinical Research Seminar
|
|
|
- Lydia Perkins
- 5 years ago
- Views:
Transcription

1 Clinical Research Seminar HOW TO DEVELOP A CORRECTIVE AND PREVENTIVE ACTION PLAN (THAT EVEN THE IRB AND FDA WILL LOVE) April 11, 2018 Fiona Rice, MPH Human Research Quality Manager fionar@bu.edu Mary-Tara Roth, RN, MSN, MPH Director, CRRO mtroth@bu.edu
2 Case Protocol requires urine dipstick test be performed at Screening; Baseline; Weeks 4, 8, 12, 16, and 24; (24 hr urine test done if dipstick protein 30 mg/dl). Dipstick is a safety assessment test drug dose may be changed, so missed protocol-required urine dipstick tests compromise subject safety. Subjects 1005 and 1018 did not have urine dipstick results recorded at baseline. Subjects 1021 and 1045 did not have urine dipstick results recorded at weeks 8 and 12 despite having elevated protein levels at week 4.
3 Objectives 1) Describe what a CAPA plan is and when it is required for studies approved by the BMC/BU Medical Campus IRB. 2) Describe the IRB process for reporting Major Deviations and Unanticipated Problems and when a CAPA is needed in the submission. 3) Describe how to develop a CAPA, including a Root Cause Analysis. 4) List elements of a strong CAPA and provide examples of insufficient CAPAs.
4 What is a CAPA? CAPA = Corrective and Preventive Action Plan Documents and communicates how a problem was fixed and controlled o Outlines REACTIVE steps needed to correct the immediate problem. o Analysis to IDENTIFY the cause(s) of the problem. o Outlines PROACTIVE STEPS needed to prevent the problem from occurring in the future.
5 When Do You Need to Develop a CAPA Plan? BMC/BU Medical Campus IRB approved Studies HRPP Policy (7.4.5) specifies that a CAPA plan must be submitted when reporting a Major Deviation (including Unanticipated Problems (UPs) that are also major deviations) IRB approved Studies with Ceded IRB Approval: WIRB/Hummingbird IRB Other IRBs (MGH, Other Universities, etc)
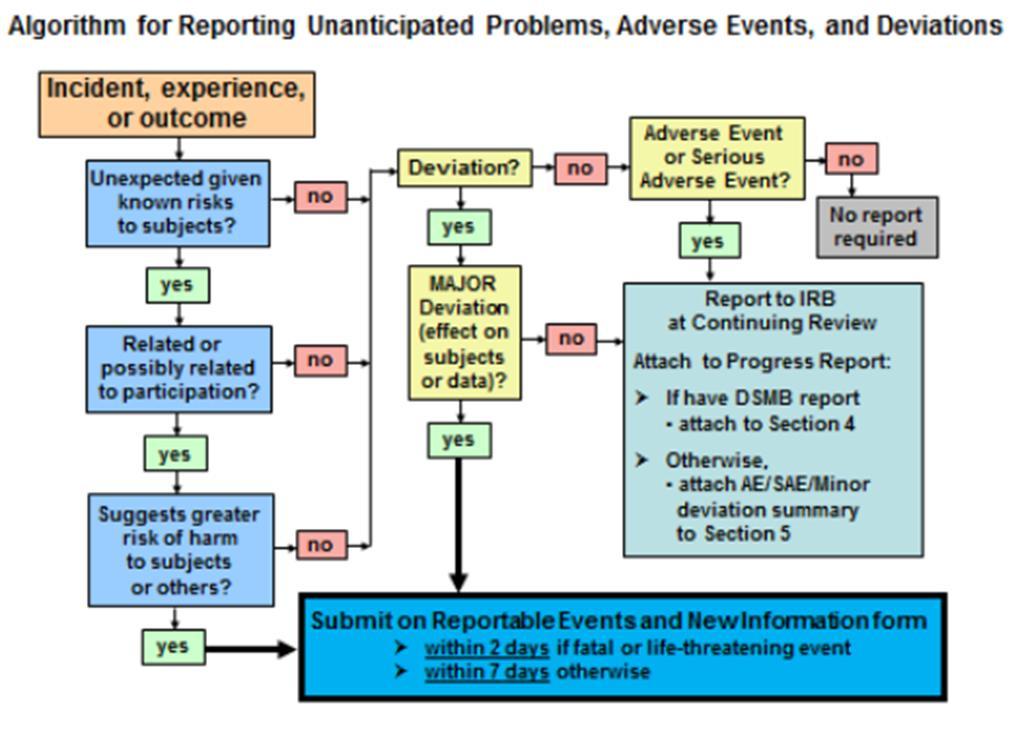
6 Major Deviations HRPP Policies and Procedures ( ) - Major Deviations Major Deviations are deviations that may (1) harm the participant's rights, safety or well-being, or (2) significantly damage the overall reliability of the study data, or (3) represent noncompliance with IRB requirements that may be serious or continuing. Major deviations must be reported to the IRB within 7 days of the investigator or research staff becoming aware of the event.
7 Unanticipated Problems OHRP considers unanticipated problems, in general, to include any incident, experience, or outcome that meets all of the following criteria: Unexpected (in terms of nature, severity, or frequency) given (a) the research procedures that are described in the protocol-related documents, such as the IRBapproved research protocol and informed consent document; and (b) the characteristics of the subject population being studied; Related or possibly related to participation in the research (meaning there is a reasonable possibility that the incident, experience, or outcome may have been caused by the procedures involved in the research); and Suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized.
8 When is a UP a Major Deviation - ora Major Deviation and UP? When the UP harms or has potential to harm participant's rights, safety or well-being, or When the UP Significantly damage the overall reliability of the study data, or When the UP Represent noncompliance with IRB requirements that may be serious or continuing.
9
10 How are Deviations and UPs Identified? Study Team/PI Observation Study Self-Audit CRRO Website has tools available ( RPN has slides available from recent self-audit presentation ( OHRA staff available for consultations (CRRO website-consultations) External Monitoring (Sponsor/ceded IRB/FDA etc.) Internal Monitoring/Auditing (OHRA routine QA Review and For-Cause Audits) Subject complaints

11 Deviations and UPs Reported to IRB via INSPIR RENI Report?
12 RENI submission Additional required information for reporting major deviations is: A description of the deviation; and Identification of whether the deviation resulted in an Unanticipated Problem (reported on the same form) Corrective and Preventive Action Plan.

13
14
15 Corrective and Preventive Action Plan (CAPA) A system for resolving quality issues Resolve problem and keep from happening again Term originating in manufacturing field CAPAs required in FDA device and pharma regulations: Quality System Regulation (21 CFR and 21 CFR 211) Clinical trials/research studies. Complex system Your product is the data Ensuring quality at every step in your study helps to ensure o o A quality product. your data Safety of subjects (and ultimately, future patients)
16 A Research study as a quality system (paraphrased from 21 CFR ) Analyze processes to identify potential causes of nonconforming product Investigate causes of nonconformities Identify the actions needed to correct/prevent problem Verify corrective and preventive actions to ensure they are effective Implement and record needed changes in methods and procedures Ensure information related to quality problems is disseminated to those responsible for assuring quality of product Submit relevant information on identified quality problems and corrective and preventive actions for management review Document activities and results

17 A Research study (or organization) as a quality system 4 main components of a quality system Say what you do Do what you say Prove it Improve it From CTTI Summary of Expert Meeting, 2010, Developing Effective Quality Systems in Clinical Trials: An Enlightened Approach
18 From FDA Guidance. The PI should ensure. A procedure for the timely correction and documentation of problems identified by study personnel, outside monitors or auditors, or other parties involved in the conduct of a study. CAPAs are the responsibility of the study PI.. FDA Guidance Investigator Responsibilities, Oct. 2009
19 Why a CAPA? Documents and communicates how the problem was fixed and controlled o Outlines REACTIVE steps needed to correct the immediate problem. o Analysis to IDENTIFY the cause(s) of the problem. o Outlines PROACTIVE STEPS steps needed to prevent the problem from occurring in the future.
20 What to do when you encounter a problem Assess potential for harm to subject(s) Notify PI Immediate corrections to protect current subject(s) Initial reporting to IRB, Sponsor, FDA, etc. (depending on seriousness) Assess causal factors (Root Cause Analysis) Implement corrective and preventive actions Further reporting to IRB, Sponsor, FDA, etc. to update if necessary Evaluate effectiveness of CAPA
21 Root Cause Analysis (RCA) Understand that clinical research studies are complex systems Often will have to assess multiple levels/processes/individuals Perform as soon as possible after the problems identified Include all individuals involved in the error
22 Root Cause Analysis You will put time, energy, possibly money into fixing the problem. You have responsibility for the rights and welfare of study participants and for the quality of the product. your data. So it s imperative to do due diligence to ensure you understand the true underlying CAUSES(S). The key to fixing and preventing problem

23 Root Cause Analysis Data Collection Causal factor charting Root cause identification Recommendation generation/implementation Evaluate implementations Rooney and Vanden Heuvel, Root Cause Analysis for Beginners, Quality Progress, July 2004
24 Root Cause Analysis Identify the problem. Review processes, interview those involved.. What happened? How did it happen? Why did it happen? When? Where? What were contributing factors? Who was involved?** Who was affected? How often did it happen? How many were affected? How serious is the problem? How extensive is the problem? Usually these questions will lead you to an underlying causal process

25 5 Whys technique State the problem: I missed a meeting at work Why 1: I left my house late Why 2: I overslept Why 3: My alarm clock didn t ring Why 4: My alarm clock was broken Why 5: My dog s tail swept my alarm clock off the nightstand during the night P. Williams, BUMC PROCEEDINGS 2001;14:
26 Fishbone diagram technique Show causes of specific problem/non-conformity Map out various levels Cause/effect F. Feldstein, Investigate; CAPA Management in Clinical Research, Sept. 2014
27 What makes a deficient CAPA? Insufficient detail on CAPAs. You have not adequately addressed how you will improve your supervision of study staff in future You did not specify the corrective actions you will take to address these violations in the future you indicate that you have the following corrective action: Investigators are required to sign a document prior to randomization that states that Inclusion Criteria have been met. Your response is inadequate because it is insufficiently detailed. You have not provided details regarding the document that investigators are required to sign and you have not submitted a copy of that document. However, although you promised certain corrective measures in your response, you did not specifically address your failure to collect the protocol-specified screening blood samples.
28 What makes a deficient CAPA? Describing corrective actions without developing SOPs Your corrective actions to ensure reporting of deaths to FDA and IRB include: reconfiguring your team, holding an IRB training for staff, informing staff that you must be notified immediately of any subject s death. We are unable to undertake an informed evaluation of your response because you did not provide documentation further explaining your corrective action plan, for example, an SOP that shows your staff is to notify you immediately upon becoming aware of any death.
29 What makes a deficient CAPA? Insufficient detail to determine if CAPA will correct the problem You indicated that you have added a clinical trials link to your site s EMR to provide access to study information for study staff. Your response is inadequate because you did not provide sufficient information to enable us to evaluate the adequacy of your corrective action plan.. it is unclear how adding a clinical trials link to your site s EMR will ensure that protocol requirements will be met for studies conducted at your site.
30 What makes a deficient CAPA? Not providing detail on corrective actions that the clinical investigator him/herself is taking We are concerned that the majority of the corrective actions appear to represent actions taken by xxx Medical Center and do not reflect corrective actions that you personally have taken.
31 What makes a deficient CAPA? Indicating that the PI doesn t understand his/her responsibilities as clinical investigator You noted that you were unaware at the time of the study that the xxx assessments for these subjects were not completed properly, and that this violation was not brought to your attention by either your study staff or sponsor monitors. We wish to emphasize that as the clinical investigator, it was your ultimate responsibility to ensure that these studies were conducted properly
32 What makes a deficient CAPA? Not providing documentation that corrective measures have been done or when they will be done Although you stated that your SOP has been put into effect and that your research coordinators are well aware of this requirement, you failed to provide documentation that your research staff have been adequately trained in this SOP.
33 What makes a deficient CAPA? Explaining how the violation occurred but not providing a corrective action

34 What makes a deficient CAPA? (a few more.) Not addressing why the problem occurred Not describing the extent/pervasiveness of the problem # times, #subjects, # studies, etc. Not detailing the timeframe of the corrective actions Not detailing how you assess if the corrective actions worked Not reporting deviations and CAPA to all applicable entities/authorities
35 CAPA - DOs A great plan may not pass IRB or FDA muster if you don t: TAKE RESPONSIBILITY APPROPRIATELY ASSESS CAUSES OF THE PROBLEM DEVELOP/MODIFY WRITTEN PROCESSES (SOPs)/PROTOCOL TRAIN STAFF ON NEW PROCESSES EVALUATE TO ENSURE YOUR CAPA WORKS DOCUMENT EVERY STEP OF YOUR CAPA Training SOPs Assessment of whether CAPA initiatives are effective
 Perform self-assessment to ensure SOP worked as part of the")
36 CAPA - DOs If you develop SOP(s) or modify the protocol as part of your CAPA Ensure the SOP addresses the root cause(s) Ensure SOP details procedures to fix and prevent the problem Train staff on this new SOP Document training (when, what, who, who) Perform self-assessment to ensure SOP worked as part of the CAPA
37 Templates to Assist you in Documentation In CRRO website under Resources go to Research and Regulatory Tools and Resources scroll to Study Regulatory Files click on Regulatory Binder Tab Inserts
38 Case revisited Protocol requires urine dipstick test be performed at Screening; Baseline; Weeks 4, 8, 12, 16, and 24; (24 hr urine test done if dipstick protein 30 mg/dl). Dipstick is a safety assessment test drug dose may be changed, so missed protocol-required urine dipstick tests compromise subject safety. Subjects 1005 and 1018 did not have urine dipstick results recorded at baseline. Subjects 1021 and 1045 did not have urine dipstick results recorded at weeks 8 and 12 despite having elevated protein levels at week 4. CAPA Plan: We have instructed our study coordinator to be more careful to ensure this dipstick test is done. What do you think?
39 Helpful guidance Investigator Responsibilities - Protecting the Rights, Safety, and Welfare of Study Subjects pdf IRB Continuing Review after Clinical Investigation Approval Oversight of Clinical Investigations - A Risk-Based Approach to Monitoring guidance pdf FDA Inspections of Clinical Investigators FDA Inspectional Objectives for CAPAs
40 Questions?
HIC Standard Operating Procedure. For-Cause Audits of Human Research Studies
 HIC Standard Operating Procedure For-Cause Audits of Human Research Studies Background As part of the Wayne State University (WSU) Human Investigation Committee s (HIC) Human Research Protection Program,
HIC Standard Operating Procedure For-Cause Audits of Human Research Studies Background As part of the Wayne State University (WSU) Human Investigation Committee s (HIC) Human Research Protection Program,
Research Audits PGR. Effective: 12/04/2013 Reviewed: 12/04/2015. Name of Associated Policy: Palmetto Health Administrative Research Review
 Effective: 12/04/2013 Reviewed: 12/04/2015 Name of Associated Policy: Palmetto Health Administrative Research Review Definitions Responsible Positions Equipment Needed Procedure Steps, Guidelines, Rules,
Effective: 12/04/2013 Reviewed: 12/04/2015 Name of Associated Policy: Palmetto Health Administrative Research Review Definitions Responsible Positions Equipment Needed Procedure Steps, Guidelines, Rules,
Clinical Trial Quality Assurance Common Findings
 Clinical Trial Quality Assurance Common Findings Objectives Identify common findings found in research study reviews conducted by the CTQA Program Understand what findings require an action plan vs. a
Clinical Trial Quality Assurance Common Findings Objectives Identify common findings found in research study reviews conducted by the CTQA Program Understand what findings require an action plan vs. a
Risk-Benefit Ratio and Determinations. Sarah Mumford, Ammon Pate, Annie Risenmay IRB Operations Managers University of Utah
 Risk-Benefit Ratio and Determinations Sarah Mumford, Ammon Pate, Annie Risenmay IRB Operations Managers University of Utah Risk-Benefit Ratio and Determinations Nuances of Risk Determinations Direct Benefit
Risk-Benefit Ratio and Determinations Sarah Mumford, Ammon Pate, Annie Risenmay IRB Operations Managers University of Utah Risk-Benefit Ratio and Determinations Nuances of Risk Determinations Direct Benefit
WIRBinar. How to Survive an FDA Inspection. Upcoming Trainings: Contact Us: (360)
") WIRBinar How to Survive an FDA Inspection 10-26-2011 Brought to you by WIRB Education and Consulting Services. Improve your ability to maintain compliance and protect human subjects with guidance from
WIRBinar How to Survive an FDA Inspection 10-26-2011 Brought to you by WIRB Education and Consulting Services. Improve your ability to maintain compliance and protect human subjects with guidance from
Dr. R. Sathianathan. Role & Responsibilities of Principal Investigators in Clinical Trials. 18 August 2015
 18 August 2015 Role & Responsibilities of Principal Dr. R. Sathianathan Professor of Psychiatry, SRMC, Porur & Former Director, Institute of Mental Health, Chennai Principal Investigators & GOOD CLINICAL
18 August 2015 Role & Responsibilities of Principal Dr. R. Sathianathan Professor of Psychiatry, SRMC, Porur & Former Director, Institute of Mental Health, Chennai Principal Investigators & GOOD CLINICAL
Biomedical IRB MS #
 Department for Human Research Protections Institutional Review Boards Biomedical IRB MS # 1035 419-383-6796 IRB.Biomed@utoledo.edu Social, Behavioral and Educational IRB MS # 944 419-530-6167 IRB.SBE@utoledo.edu
Department for Human Research Protections Institutional Review Boards Biomedical IRB MS # 1035 419-383-6796 IRB.Biomed@utoledo.edu Social, Behavioral and Educational IRB MS # 944 419-530-6167 IRB.SBE@utoledo.edu
Study Management SM STANDARD OPERATING PROCEDURE FOR Adverse Event Reporting
 Study Management SM 306.00 STANDARD OPERATING PROCEDURE FOR Adverse Event Reporting Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick M. Schnell, MD,
Study Management SM 306.00 STANDARD OPERATING PROCEDURE FOR Adverse Event Reporting Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick M. Schnell, MD,
University of South Carolina. Unanticipated Problems and Adverse Events Guidelines
 University of South Carolina Unanticipated Problems and Adverse Events Guidelines These guidelines define the procedures of USC for addressing unanticipated problems involving risks to research participants
University of South Carolina Unanticipated Problems and Adverse Events Guidelines These guidelines define the procedures of USC for addressing unanticipated problems involving risks to research participants
EFFECTIVE ROOT CAUSE ANALYSIS AND CORRECTIVE ACTION PROCESS
 I International Symposium Engineering Management And Competitiveness 2011 (EMC2011) June 24-25, 2011, Zrenjanin, Serbia EFFECTIVE ROOT CAUSE ANALYSIS AND CORRECTIVE ACTION PROCESS Branislav Tomić * Senior
I International Symposium Engineering Management And Competitiveness 2011 (EMC2011) June 24-25, 2011, Zrenjanin, Serbia EFFECTIVE ROOT CAUSE ANALYSIS AND CORRECTIVE ACTION PROCESS Branislav Tomić * Senior
Public Input for Changes to Reportable Events Policy
 Public Input for Changes to Reportable Events Policy May 23, 2017 Richard Guido, MD, IRB Chair Jamie Zelazny, PhD, RN, Regulatory Affairs Specialist Outline Regulatory basis for reporting policies Importance
Public Input for Changes to Reportable Events Policy May 23, 2017 Richard Guido, MD, IRB Chair Jamie Zelazny, PhD, RN, Regulatory Affairs Specialist Outline Regulatory basis for reporting policies Importance
Reporting to the IRB How to Report the Essentials and Improve the Protection of Human Subjects
 Webinar Series Reporting to the IRB How to Report the Essentials and Improve the Protection of Human Subjects April 10, 2013 Presented by: James MacFarlane Director of Board Operations About the Webinar
Webinar Series Reporting to the IRB How to Report the Essentials and Improve the Protection of Human Subjects April 10, 2013 Presented by: James MacFarlane Director of Board Operations About the Webinar
When a Single IRB Reviews for Multiple Sites:
 When a Single IRB Reviews for Multiple Sites: The Complexities of Simplification John Ennever, MD, PhD Director Office of Human Research Affairs Boston Medical Center and Boston University Medical Campus
When a Single IRB Reviews for Multiple Sites: The Complexities of Simplification John Ennever, MD, PhD Director Office of Human Research Affairs Boston Medical Center and Boston University Medical Campus
I. Scope This policy defines unanticipated problems and adverse events and establishes the reporting process and timeline.
 Human Research Protection Program Policies & Procedures Unanticipated Problems and Adverse Events Version 3.0 Date Effective: 11.9.2012 Research Integrity Office Mail code L106-RI Portland, Oregon 97239-3098
Human Research Protection Program Policies & Procedures Unanticipated Problems and Adverse Events Version 3.0 Date Effective: 11.9.2012 Research Integrity Office Mail code L106-RI Portland, Oregon 97239-3098
Quality Assurance and Performance Improvement (QAPI)
") Quality Assurance and Performance Improvement () Carol Hill, MSN, RN, RAC-MT, DNS-CT, QCP-MT, CPC Objectives Identify the 5 key elements that form the framework of a program Recognize process tools that
Quality Assurance and Performance Improvement () Carol Hill, MSN, RN, RAC-MT, DNS-CT, QCP-MT, CPC Objectives Identify the 5 key elements that form the framework of a program Recognize process tools that
Clinical Research Resources Office ReSPECT Registry & StudyFinder at BU/BMC
 Clinical Research Resources Office ReSPECT Registry & StudyFinder at BU/BMC Farrah Belizaire, BS R. Joshua Reynolds, BM Mary-Tara Roth, RN, MSN, MPH Clinical Research Resources Office (CRRO) November 14,
Clinical Research Resources Office ReSPECT Registry & StudyFinder at BU/BMC Farrah Belizaire, BS R. Joshua Reynolds, BM Mary-Tara Roth, RN, MSN, MPH Clinical Research Resources Office (CRRO) November 14,
UNIVERSITY OF TENNESSEE HEALTH SCIENCE CENTER INSTITUTIONAL REVIEW BOARD REPORTING UNANTICIPATED PROBLEMS INCLUDING ADVERSE EVENTS
 UNIVERSITY OF TENNESSEE HEALTH SCIENCE CENTER INSTITUTIONAL REVIEW BOARD REPORTING UNANTICIPATED PROBLEMS INCLUDING ADVERSE EVENTS I. PURPOSE To specify the procedures for reporting unanticipated problems,
UNIVERSITY OF TENNESSEE HEALTH SCIENCE CENTER INSTITUTIONAL REVIEW BOARD REPORTING UNANTICIPATED PROBLEMS INCLUDING ADVERSE EVENTS I. PURPOSE To specify the procedures for reporting unanticipated problems,
NN SS 401 NEURONEXT NETWORK STANDARD OPERATING PROCEDURE FOR SITE SELECTION AND QUALIFICATION
 NN SS 401 NEURONEXT NETWORK STANDARD OPERATING PROCEDURE FOR SITE SELECTION AND QUALIFICATION SOP: NN SS 401 Version No.: 2.0 Effective Date: 21Oct2016 SITE SELECTION AND QUALIFICATION Supercedes Document:
NN SS 401 NEURONEXT NETWORK STANDARD OPERATING PROCEDURE FOR SITE SELECTION AND QUALIFICATION SOP: NN SS 401 Version No.: 2.0 Effective Date: 21Oct2016 SITE SELECTION AND QUALIFICATION Supercedes Document:
University of Colorado Denver Human Research Protection Program Investigator Responsibilities for the Protection of Human Subjects
 Institutional Guidelines The Colorado Multiple Institutional Review Board (COMIRB) recently reviewed and approved your research. The COMIRB reviews research to ensure that the federal regulations for protecting
Institutional Guidelines The Colorado Multiple Institutional Review Board (COMIRB) recently reviewed and approved your research. The COMIRB reviews research to ensure that the federal regulations for protecting
PROMPTLY REPORTABLE EVENTS
 PROMPTLY REPORTABLE EVENTS PURPOSE AND SCOPE To define the structure and responsibility for reporting unanticipated problems that occurs during the conduct of research. APPLICABLE REGULATIONS Policy II.02
PROMPTLY REPORTABLE EVENTS PURPOSE AND SCOPE To define the structure and responsibility for reporting unanticipated problems that occurs during the conduct of research. APPLICABLE REGULATIONS Policy II.02
12.0 Investigator Responsibilities
 12.0 Investigator Responsibilities 12.1 Policy Investigators are ultimately responsible for the conduct of research. Research must be conducted according to the signed Investigator statement, the investigational
12.0 Investigator Responsibilities 12.1 Policy Investigators are ultimately responsible for the conduct of research. Research must be conducted according to the signed Investigator statement, the investigational
SOP : Quality Assurance Inspections SCOPE RESPONSIBILITIES. APPROVAL AUTHORITY EFFECTIVE DATE May PURPOSE 2.
 TITLE SCOPE RESPONSIBILITIES APPROVAL AUTHORITY EFFECTIVE DATE May 2018 901: Quality Assurance Inspections All research submitted to the University of British Columbia s Research Ethics Boards The Vice
TITLE SCOPE RESPONSIBILITIES APPROVAL AUTHORITY EFFECTIVE DATE May 2018 901: Quality Assurance Inspections All research submitted to the University of British Columbia s Research Ethics Boards The Vice
QUALITY TIPS FOR CLINICAL SITES. Athena Thomas-Visel. Clinical Quality Consultant QUALITY TIPS FOR CLINICAL SITES
 QUALITY TIPS FOR CLINICAL SITES Athena Thomas-Visel Clinical Quality Consultant QUALITY TIPS FOR CLINICAL SITES Purpose of presentation: Share best practices seen from 150+ sites visited Spark conversation
QUALITY TIPS FOR CLINICAL SITES Athena Thomas-Visel Clinical Quality Consultant QUALITY TIPS FOR CLINICAL SITES Purpose of presentation: Share best practices seen from 150+ sites visited Spark conversation
SOP Problems and Adverse Events, Record and Report
 Office of Research Integrity - Human Subjects SOP #: ORI(HS)- 1.0 Page #: Page 1 of 5 Approved By: ORI Executive Director *Signature on file Date: Date First Effective: 11/18/2013 Approved by: Biomedical
Office of Research Integrity - Human Subjects SOP #: ORI(HS)- 1.0 Page #: Page 1 of 5 Approved By: ORI Executive Director *Signature on file Date: Date First Effective: 11/18/2013 Approved by: Biomedical
Keele Clinical Trials Unit
 Keele Clinical Trials Unit Standard Operating Procedure (SOP) Summary Box Title SOP Index Number SOP 21 Version 4.0 Approval Date Effective Date Non-Compliance: Deviations and Serious Breaches of GCP and/or
Keele Clinical Trials Unit Standard Operating Procedure (SOP) Summary Box Title SOP Index Number SOP 21 Version 4.0 Approval Date Effective Date Non-Compliance: Deviations and Serious Breaches of GCP and/or
Standard Operating Procedures
 Clinical Monitoring and Site Verification Procedure Overview To define the standard procedures for preparation and documentation of site visits for clinical monitoring and spoke verification for any NETT
Clinical Monitoring and Site Verification Procedure Overview To define the standard procedures for preparation and documentation of site visits for clinical monitoring and spoke verification for any NETT
Connie Hoy October 2013
 Connie Hoy October 2013 Warning letter issued to firm for complaint handling Failure to report within 30 days Failure to implement adequate procedures Failure to establish standard review process No procedure
Connie Hoy October 2013 Warning letter issued to firm for complaint handling Failure to report within 30 days Failure to implement adequate procedures Failure to establish standard review process No procedure
National Health Regulatory Authority Kingdom of Bahrain
 National Health Regulatory Authority Kingdom of Bahrain THE NHRA GUIDANCE ON SERIOUS ADVERSE EVENT MANAGEMENT AND REPORTING THE PURPOSE OF THIS DOCUMENT IS TO OUTLINE SERIOUS ADVERSE EVENTS THAT SHOULD
National Health Regulatory Authority Kingdom of Bahrain THE NHRA GUIDANCE ON SERIOUS ADVERSE EVENT MANAGEMENT AND REPORTING THE PURPOSE OF THIS DOCUMENT IS TO OUTLINE SERIOUS ADVERSE EVENTS THAT SHOULD
Investigator-Initiated Studies: When you re the Sponsor. Cheri Robert & Tammy Mah-Fraser
 Investigator-Initiated Studies: When you re the Sponsor Cheri Robert & Tammy Mah-Fraser ACRC Clinical Research Conference Edmonton, AB October 15, 2014 Session Objectives Define roles of the investigator,
Investigator-Initiated Studies: When you re the Sponsor Cheri Robert & Tammy Mah-Fraser ACRC Clinical Research Conference Edmonton, AB October 15, 2014 Session Objectives Define roles of the investigator,
Documenting the Story of a Clinical Trial: Concept to CAPA. Lori T. Gilmartin Gilmartin Consulting LLC
 Documenting the Story of a Clinical Trial: Concept to CAPA Lori T. Gilmartin Gilmartin Consulting LLC The regulations represent the floor while ethical thinking is the sky. Dr. Thomas Moore Boston University
Documenting the Story of a Clinical Trial: Concept to CAPA Lori T. Gilmartin Gilmartin Consulting LLC The regulations represent the floor while ethical thinking is the sky. Dr. Thomas Moore Boston University
Quality Assurance and Site Monitoring Visits. Introduction. Training Outline
 Instructors: 2009 Web Seminar Series Quality Assurance and Site Monitoring Visits Amanda Moore and Scott Provost, M.M., M.S.W. "This training has been funded in whole or in part with Federal funds from
Instructors: 2009 Web Seminar Series Quality Assurance and Site Monitoring Visits Amanda Moore and Scott Provost, M.M., M.S.W. "This training has been funded in whole or in part with Federal funds from
The GCP Perspective on Study Monitoring
 The GCP Perspective on Study Monitoring Heidi Judge, CCRP Sr. Clinical Trials Project Manager Clinical Trials Network and Institute Massachusetts General Hospital 1 Overview Monitoring Basics Who, What,
The GCP Perspective on Study Monitoring Heidi Judge, CCRP Sr. Clinical Trials Project Manager Clinical Trials Network and Institute Massachusetts General Hospital 1 Overview Monitoring Basics Who, What,
Title: Investigator Responsibilities. SOP Number: 1501 Effective Date: June 2, 2017
 Previous Version Dates: Title: Investigator Responsibilities SOP Number: 1501 Effective Date: June 2, 2017 1 Purpose Investigators are ultimately responsible for the conduct of research. Investigators
Previous Version Dates: Title: Investigator Responsibilities SOP Number: 1501 Effective Date: June 2, 2017 1 Purpose Investigators are ultimately responsible for the conduct of research. Investigators
General Administration GA STANDARD OPERATING PROCEDURE FOR Document Development and Change Control
 General Administration GA 104.00 STANDARD OPERATING PROCEDURE FOR Document Development and Change Control Approval: Nancy Paris, MS, FACHE President and CEO 08 March 2012 (Signature and Date) Approval:
General Administration GA 104.00 STANDARD OPERATING PROCEDURE FOR Document Development and Change Control Approval: Nancy Paris, MS, FACHE President and CEO 08 March 2012 (Signature and Date) Approval:
4.2. Clinical Trial Monitor (or Monitor): The person responsible for monitoring the data on behalf of the sponsor or contract research organization.
: The person responsible for monitoring the data on behalf of the sponsor or contract research organization.") SOP #: MON-101 Page: 1 of 6 1. POLICY STATEMENT: The DF/HCC understands that external sponsors are required to monitor the progress of clinical investigations and ensure appropriate research data collection
SOP #: MON-101 Page: 1 of 6 1. POLICY STATEMENT: The DF/HCC understands that external sponsors are required to monitor the progress of clinical investigations and ensure appropriate research data collection
TITLE: Reporting Adverse Events SOP #: RCO-204 Page: 1 of 5 Effective Date: 01/31/18
 SOP #: RCO-204 Page: 1 of 5 1. POLICY STATEMENT: The research team is responsible for recognizing changes in subject health that may qualify as adverse events, classifying those results as defined in the
SOP #: RCO-204 Page: 1 of 5 1. POLICY STATEMENT: The research team is responsible for recognizing changes in subject health that may qualify as adverse events, classifying those results as defined in the
Archived. DPC: Corrective Action. Quality Manual
 actions 4.9.2 Levels of nonconformity 4.9.1.c 4.9.1.d 4.11. Laboratories may experience technical or administrative nonconformities. These occurrences can be adverse to the quality of the work product
actions 4.9.2 Levels of nonconformity 4.9.1.c 4.9.1.d 4.11. Laboratories may experience technical or administrative nonconformities. These occurrences can be adverse to the quality of the work product
INSPIRing Changes to the IRB Process: New templates and more
 INSPIRing Changes to the IRB Process: New templates and more John F. Ennever, MD, PhD, CIP Director, Human Research Protection Program Office of Human Research Affairs Boston Medical Center and Boston
INSPIRing Changes to the IRB Process: New templates and more John F. Ennever, MD, PhD, CIP Director, Human Research Protection Program Office of Human Research Affairs Boston Medical Center and Boston
STUDY INFORMATION POST-IRB APPROVAL FDA DEVICE (IDE) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR 812)
 SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR 812)") POST-IRB APPROVAL FDA DEVICE (IDE) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR 812) Purpose: Investigators who initiate and submit an IDE application to the FDA assume the responsibilities of both
POST-IRB APPROVAL FDA DEVICE (IDE) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR 812) Purpose: Investigators who initiate and submit an IDE application to the FDA assume the responsibilities of both
Review of F323 Related to Falls. Marilyn Hirsch Region V December 16, 2015
 Review of F323 Related to Falls Marilyn Hirsch Region V December 16, 2015 Objectives Describe Region V F323 Project Review F323 Regulation Review and discuss citations History At our meeting two years
Review of F323 Related to Falls Marilyn Hirsch Region V December 16, 2015 Objectives Describe Region V F323 Project Review F323 Regulation Review and discuss citations History At our meeting two years
Good Clinical Practice: A Ground Level View
 Good Clinical Practice: A Ground Level View Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Clinical Research Administration Research Institute,
Good Clinical Practice: A Ground Level View Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Clinical Research Administration Research Institute,
Solutions for GCP Compliance Challenges. September 23, 2015 Northwestern University IRB Brown Bag Session
 Solutions for GCP Compliance Challenges September 23, 2015 Northwestern University IRB Brown Bag Session PAMELA MASON, MPH Vice President, Mason Professional Services, LLC Adjunct Faculty, Northwestern
Solutions for GCP Compliance Challenges September 23, 2015 Northwestern University IRB Brown Bag Session PAMELA MASON, MPH Vice President, Mason Professional Services, LLC Adjunct Faculty, Northwestern
Solutions for GCP Compliance Challenges
 Solutions for GCP Compliance Challenges September 23, 2015 Northwestern University IRB Brown Bag Session PAMELA MASON, MPH Vice President, Mason Professional Services, LLC Adjunct Faculty, Northwestern
Solutions for GCP Compliance Challenges September 23, 2015 Northwestern University IRB Brown Bag Session PAMELA MASON, MPH Vice President, Mason Professional Services, LLC Adjunct Faculty, Northwestern
Margaret Huber, RN, CHRC Compliance Consultant Office of Research Compliance
 Margaret Huber, RN, CHRC Compliance Consultant Office of Research Compliance 4/20/2015 Objectives Define monitoring and explain why monitoring is important in clinical trials Provide an overview of the
Margaret Huber, RN, CHRC Compliance Consultant Office of Research Compliance 4/20/2015 Objectives Define monitoring and explain why monitoring is important in clinical trials Provide an overview of the
General Administration GA STANDARD OPERATING PROCEDURE FOR Sponsor Responsibility and Delegation of Responsibility
 General Administration GA 102.01 STANDARD OPERATING PROCEDURE FOR Sponsor Responsibility and Delegation of Responsibility Approval: Nancy Paris, MS, FACHE President and CEO (17 July 2014) (Signature and
General Administration GA 102.01 STANDARD OPERATING PROCEDURE FOR Sponsor Responsibility and Delegation of Responsibility Approval: Nancy Paris, MS, FACHE President and CEO (17 July 2014) (Signature and
DEPARTMENT OF HEALTH & HUMAN SERVICES WARNING LETTER. (b) (4) clinical investigation (Protocol entitled A Phase II, Multicenter,
 (4) clinical investigation (Protocol entitled A Phase II, Multicenter,") DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service Food and Drug Administration Rockville, MD 20857 WARNING LETTER CERTIFIED MAIL RETURN RECEIPT REQUESTED Ronald Bukowski, M.D. 28099 Gates Mills
DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service Food and Drug Administration Rockville, MD 20857 WARNING LETTER CERTIFIED MAIL RETURN RECEIPT REQUESTED Ronald Bukowski, M.D. 28099 Gates Mills
SAINT AGNES MEDICAL CENTER CLINICAL RESEARCH CENTER Fresno, California. STANDARD OPERATING PROCEDURES Institutional Review Board
 SAINT AGNES MEDICAL CENTER CLINICAL RESEARCH CENTER Fresno, California STANDARD OPERATING PROCEDURES Institutional Review Board Date Effective: April 26, 2001 Index No. R 1217 Date Last Revised: 0 Date
SAINT AGNES MEDICAL CENTER CLINICAL RESEARCH CENTER Fresno, California STANDARD OPERATING PROCEDURES Institutional Review Board Date Effective: April 26, 2001 Index No. R 1217 Date Last Revised: 0 Date
Good Documentation Practices. Human Subject Research. for
 Good Documentation Practices for Human Subject Research Bridget M. Psicihulis, RHIA, CCRC Quality Improvement Unit Coordinator Human Research Protection Program Wheaton Franciscan Healthcare (last updated
Good Documentation Practices for Human Subject Research Bridget M. Psicihulis, RHIA, CCRC Quality Improvement Unit Coordinator Human Research Protection Program Wheaton Franciscan Healthcare (last updated
Mastering Clinical Research April 19, :30 am
 Mastering Clinical Research April 19, 2017 7:30 am New Question and Answer Response System Log In Directions Use the following link to access pre and post test questions: http://www.socrative.com/ Click
Mastering Clinical Research April 19, 2017 7:30 am New Question and Answer Response System Log In Directions Use the following link to access pre and post test questions: http://www.socrative.com/ Click
Overview ICH GCP E6(R2) Integrated Addendum
 Integrated Addendum") 2017 Biomedical Research Alliance of New York LLC CITI Program is a division of BRANY Overview ICH GCP E6(R2) Integrated Addendum Introduction On 15 December 2016, the International Council for Harmonistion
2017 Biomedical Research Alliance of New York LLC CITI Program is a division of BRANY Overview ICH GCP E6(R2) Integrated Addendum Introduction On 15 December 2016, the International Council for Harmonistion
Local VA VA ORD CSP Other VA ORD. IRB of Record Registration Number: IRB Operated by: Local VA Non-local VA Academic Affiliate VHA Central IRB
 ADMINISTRATIVE INFORMATION Principal Investigator: Study Coordinator: Protocol Title: Current Audit Date: Research Compliance Officer (RCO) or Designated Auditor(s): Local VA VA ORD CSP Other VA ORD Sponsor
ADMINISTRATIVE INFORMATION Principal Investigator: Study Coordinator: Protocol Title: Current Audit Date: Research Compliance Officer (RCO) or Designated Auditor(s): Local VA VA ORD CSP Other VA ORD Sponsor
Building Quality into Clinical Trials. Amy C. Hoeper, MSN, RN, CCRC, Quality Manager Cincinnati Children s Gamble Program for Clinical Studies
 Building Quality into Clinical Trials Amy C. Hoeper, MSN, RN, CCRC, Quality Manager Cincinnati Children s Gamble Program for Clinical Studies Objectives Identify strategies for developing a Quality Management
Building Quality into Clinical Trials Amy C. Hoeper, MSN, RN, CCRC, Quality Manager Cincinnati Children s Gamble Program for Clinical Studies Objectives Identify strategies for developing a Quality Management
MANAGEMENT OF PROTOCOL AND GCP DEVIATIONS AND VIOLATIONS
 MANAGEMENT OF PROTOCOL AND GCP DEVIATIONS AND VIOLATIONS DOCUMENT NO.: CR010 v4.0 AUTHOR: Heather Charles ISSUE DATE: 01 September 2016 EFFECTIVE DATE: 15 September 2016 1 INTRODUCTION 1.1 The Academic
MANAGEMENT OF PROTOCOL AND GCP DEVIATIONS AND VIOLATIONS DOCUMENT NO.: CR010 v4.0 AUTHOR: Heather Charles ISSUE DATE: 01 September 2016 EFFECTIVE DATE: 15 September 2016 1 INTRODUCTION 1.1 The Academic
Medical Director Requirements for Nursing Facilities Advance Issuance of Revised Survey Guidance HIGHLIGHTS
 Medical Director Requirements for Nursing Facilities Advance Issuance of Revised Survey Guidance HIGHLIGHTS On April 9, 2005 the Centers for Medicare and Medicaid Services (CMS) released revised interpretive
Medical Director Requirements for Nursing Facilities Advance Issuance of Revised Survey Guidance HIGHLIGHTS On April 9, 2005 the Centers for Medicare and Medicaid Services (CMS) released revised interpretive
Request to Use an External IRB as an IRB of Record
 This form is to be used by investigators requesting use of an external IRB. Please submit this completed form, along with the required attachments, to the MHC IRB at hrpp@mclaren.org. (Please see SOP:
This form is to be used by investigators requesting use of an external IRB. Please submit this completed form, along with the required attachments, to the MHC IRB at hrpp@mclaren.org. (Please see SOP:
Theradex Audit 2013: Findings & Corrective Action
 Theradex Audit 2013: Findings & Corrective Action Overview Discuss Findings and CAP for: Informed Consent Content IRB Informed Consent Eligibility Treatment Serious Adverse Events Response General Data
Theradex Audit 2013: Findings & Corrective Action Overview Discuss Findings and CAP for: Informed Consent Content IRB Informed Consent Eligibility Treatment Serious Adverse Events Response General Data
SOP-QA-28 V2. Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head of School
 Title: Effective Date: 1-4-17 Review Date: 1-4-20 Author: Richard Cowie, QA Manager QA Approval: Richard Cowie, QA Manager Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head
Title: Effective Date: 1-4-17 Review Date: 1-4-20 Author: Richard Cowie, QA Manager QA Approval: Richard Cowie, QA Manager Approver: Prof Maggie Cruickshank, R&D Director Approver: Prof Steve Heys, Head
How to Prepare for Federal Inspections and What to Expect
 How to Prepare for Federal Inspections and What to Expect Jennifer A. Graf Tufts Medical Center Tufts University Health Sciences Director of IRB Operations Originally presented at the February 2011 Society
How to Prepare for Federal Inspections and What to Expect Jennifer A. Graf Tufts Medical Center Tufts University Health Sciences Director of IRB Operations Originally presented at the February 2011 Society
Standard Operating Procedure Research Governance
 Research and Enterprise Standard Operating Procedure Research Governance Title: Research Governance Audit SOP Reference Number: QUB-ADRE-08 Date prepared 7 August 008 Version Number: Final v -6.0 Revision
Research and Enterprise Standard Operating Procedure Research Governance Title: Research Governance Audit SOP Reference Number: QUB-ADRE-08 Date prepared 7 August 008 Version Number: Final v -6.0 Revision
WARNING LETTER CERTIFIED MAIL RETURN RECEIPT REQUESTED
 DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service Food and Drug Administration Rockville, MD 20857 WARNING LETTER CERTIFIED MAIL RETURN RECEIPT REQUESTED Charles J. Coté, M.D. Ref: 09-HFD-45-02-04
DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service Food and Drug Administration Rockville, MD 20857 WARNING LETTER CERTIFIED MAIL RETURN RECEIPT REQUESTED Charles J. Coté, M.D. Ref: 09-HFD-45-02-04
Corrective and Preventive Action
 QP 15.0 Corrective and Preventive Action Contents 1.0 Scope 1.1 General 1.2 References 1.3 Responsibilities 1.4 Definitions 1.5 Approvals 2.0 Procedures 2.1 Complaint Handling 2.2 Corrective and Preventive
QP 15.0 Corrective and Preventive Action Contents 1.0 Scope 1.1 General 1.2 References 1.3 Responsibilities 1.4 Definitions 1.5 Approvals 2.0 Procedures 2.1 Complaint Handling 2.2 Corrective and Preventive
Postmarketing Drug Safety and Inspection Readiness
 Postmarketing Drug Safety and Inspection Readiness June 19, 2018 Center for Drug Evaluation and Research (CDER) Small Business and Industry Assistance (SBIA) Webinar United States Food and Drug Administration
Postmarketing Drug Safety and Inspection Readiness June 19, 2018 Center for Drug Evaluation and Research (CDER) Small Business and Industry Assistance (SBIA) Webinar United States Food and Drug Administration
FDA Medical Device Regulations vs. ISO 14155
 Vol. 11, No. 9, September 2015 Happy Trials to You FDA Medical Device Regulations vs. ISO 14155 By Shawn Kennedy Medical device clinical trials must comply with 21 CFR Parts 11 (Electronic Records), 50
Vol. 11, No. 9, September 2015 Happy Trials to You FDA Medical Device Regulations vs. ISO 14155 By Shawn Kennedy Medical device clinical trials must comply with 21 CFR Parts 11 (Electronic Records), 50
Investigator Roles and Responsibilities in Clinical Device Trials
 Investigator Roles and Responsibilities in Clinical Device Trials A Total Product Lifecycle Approach to Medical Device Development: Responsibilities and Opportunities The Stanford Center for Clinical and
Investigator Roles and Responsibilities in Clinical Device Trials A Total Product Lifecycle Approach to Medical Device Development: Responsibilities and Opportunities The Stanford Center for Clinical and
AN OVERVIEW OF CLINICAL STUDY TASKS AND ACTIVITIES
 1 AN OVERVIEW OF CLINICAL STUDY TASKS AND ACTIVITIES Key Clinical Study Tasks and Activities 2 Discussion of Key Tasks and Activities 3 Development of the Clinical Protocol and Study Materials 3 Qualification
1 AN OVERVIEW OF CLINICAL STUDY TASKS AND ACTIVITIES Key Clinical Study Tasks and Activities 2 Discussion of Key Tasks and Activities 3 Development of the Clinical Protocol and Study Materials 3 Qualification
20 STEPS FROM STUDY IDEA INCEPTION TO PUBLISHING RESEARCH/ Evidence-Based Practice
 20 STEPS FROM STUDY IDEA INCEPTION TO PUBLISHING RESEARCH/ Evidence-Based Practice Nursing Research/ Evidence-Based Practice Checklist (Version 31 January 2012) Specify the date in the left column when
20 STEPS FROM STUDY IDEA INCEPTION TO PUBLISHING RESEARCH/ Evidence-Based Practice Nursing Research/ Evidence-Based Practice Checklist (Version 31 January 2012) Specify the date in the left column when
Office of the Vice Chancellor for Research Supervisory Responsibilities of Clinical Investigators
 Office of the Vice Chancellor for Research Supervisory Responsibilities of Clinical Investigators Patricia Fischer, RN, CCRP Investigator Responsibilities FDA Draft Guidance May 2007 Clarifies FDA s expectations,
Office of the Vice Chancellor for Research Supervisory Responsibilities of Clinical Investigators Patricia Fischer, RN, CCRP Investigator Responsibilities FDA Draft Guidance May 2007 Clarifies FDA s expectations,
ONADE s Data Quality Review
 ONADE s Data Quality Review Office of New Animal Drug Evaluation Center for Veterinary Medicine Regulatory Affairs in Animal Health Seminar Kansas State University Olathe March 06, 2018 Presenter: Ana
ONADE s Data Quality Review Office of New Animal Drug Evaluation Center for Veterinary Medicine Regulatory Affairs in Animal Health Seminar Kansas State University Olathe March 06, 2018 Presenter: Ana
MARKEY CANCER CENTER CLINICAL RESEARCH ORGANIZATION STANDARD OPERATING PROCEDURES SOP No.: MCCCRO-D
 Page 1 of 8 MARKEY CANCER CENTER CLINICAL RESEARCH ORGANIZATION STANDARD OPERATING PROCEDURES SOP No.: Title: Data Safety and Monitoring Committee Administrative and Revision: N/A Revision Date: N/A Functional
Page 1 of 8 MARKEY CANCER CENTER CLINICAL RESEARCH ORGANIZATION STANDARD OPERATING PROCEDURES SOP No.: Title: Data Safety and Monitoring Committee Administrative and Revision: N/A Revision Date: N/A Functional
IDENTIFYING, RECORDING AND REPORTING ADVERSE EVENTS FOR CLINICAL INVESTIGATIONS OF MEDICAL DEVICES
 IDENTIFYING, RECORDING AND REPORTING ADVERSE EVENTS FOR CLINICAL INVESTIGATIONS OF MEDICAL DEVICES DOCUMENT NO.: CR012 v2.0 AUTHOR: Raymond French ISSUE DATE: 18 September 2017 EFFECTIVE DATE: 02 October
IDENTIFYING, RECORDING AND REPORTING ADVERSE EVENTS FOR CLINICAL INVESTIGATIONS OF MEDICAL DEVICES DOCUMENT NO.: CR012 v2.0 AUTHOR: Raymond French ISSUE DATE: 18 September 2017 EFFECTIVE DATE: 02 October
BIMO SITE AUDIT CHECKLIST
 Item AUTHORITY AND ADMINISTRATION FOR STUDIES INVOLVING HUMAN DRUGS, BIOLOGICS AND DEVICES 1. Compare the Investigator Agreement with the information provided by the assigning Center. Auditor will check
Item AUTHORITY AND ADMINISTRATION FOR STUDIES INVOLVING HUMAN DRUGS, BIOLOGICS AND DEVICES 1. Compare the Investigator Agreement with the information provided by the assigning Center. Auditor will check
September 2-3, 2013 Chengdu, China
 Root Cause Investigations and Corrective Actions for GCP Compliance September 2-3, 2013 Chengdu, China Good Clinical Practice (GCP) is a compilation of best practices and quality standards to be applied
Root Cause Investigations and Corrective Actions for GCP Compliance September 2-3, 2013 Chengdu, China Good Clinical Practice (GCP) is a compilation of best practices and quality standards to be applied
DO I NEED TO SUBMIT FOR THIS?... & OTHER FREQUENTLY ASKED QUESTIONS. March 2015 IRB Forum
 DO I NEED TO SUBMIT FOR THIS?... & OTHER FREQUENTLY ASKED QUESTIONS March 2015 IRB Forum Topics Quality Assurance/Quality Improvement Projects Informed Consent- when is a waiver appropriate? Retrospective/Prospective
DO I NEED TO SUBMIT FOR THIS?... & OTHER FREQUENTLY ASKED QUESTIONS March 2015 IRB Forum Topics Quality Assurance/Quality Improvement Projects Informed Consent- when is a waiver appropriate? Retrospective/Prospective
SARASOTA MEMORIAL HOSPITAL CANCER RESEARCH PROGRAM POLICY
 PS1006 SARASOTA MEMORIAL HOSPITAL CANCER RESEARCH PROGRAM POLICY TITLE: Satellite Site Management Plan Job Title of Reviewer: POLICY #: EFFECTIVE DATE: REVISED DATE: POLICY TYPE: Elizabeth Carr, R.N.,
PS1006 SARASOTA MEMORIAL HOSPITAL CANCER RESEARCH PROGRAM POLICY TITLE: Satellite Site Management Plan Job Title of Reviewer: POLICY #: EFFECTIVE DATE: REVISED DATE: POLICY TYPE: Elizabeth Carr, R.N.,
UConn Health Office of Clinical & Translational Research Standard Operating Procedures
 Title: Forms & Templates Research Financial Compliance Monitoring Program Relates to Policy/Procedures: 2006-12 SOP#: 802-09 Version 7.0 Prepared by: Judie Fine Original date: 7/30/09 Approved by: Judi
Title: Forms & Templates Research Financial Compliance Monitoring Program Relates to Policy/Procedures: 2006-12 SOP#: 802-09 Version 7.0 Prepared by: Judie Fine Original date: 7/30/09 Approved by: Judi
Self-Monitoring Tool
 This form is designed for research personnel to use to assess their compliance with TTUHSC El Paso IRB policies and procedures, and federal regulations and guidance governing research with human subjects,
This form is designed for research personnel to use to assess their compliance with TTUHSC El Paso IRB policies and procedures, and federal regulations and guidance governing research with human subjects,
SOP:14:QA:110:01:NIBT PAGE 1 of 8
 SOP:14:QA:110:01:NIBT PAGE 1 of 8 Northern Ireland Blood Transfusion Service STANDARD OPERATING PROCEDURE (Operational Copy) Document Details Document Number: SOP:14:QA:110:01:NIBT Supersedes Number: Not
SOP:14:QA:110:01:NIBT PAGE 1 of 8 Northern Ireland Blood Transfusion Service STANDARD OPERATING PROCEDURE (Operational Copy) Document Details Document Number: SOP:14:QA:110:01:NIBT Supersedes Number: Not
CLOSE OUT VISIT REPORT (NO CRF TO MONITOR)
") Date: Page: 1 of 8 CLOSE OUT VISIT REPORT (NO CRF TO MONITOR) Protocol: PI Name: PI Address: Date of Visit: Monitor(s): Other Sponsor Personnel Present: Site Personnel Present at Visit (include names and
Date: Page: 1 of 8 CLOSE OUT VISIT REPORT (NO CRF TO MONITOR) Protocol: PI Name: PI Address: Date of Visit: Monitor(s): Other Sponsor Personnel Present: Site Personnel Present at Visit (include names and
QUALITY ASSURANCE PROGRAM
 QUALITY ASSURANCE PROGRAM Elaine Armstrong, MS Quality Assurance Manager PURPOSE Verify accuracy of submitted data Verify compliance with protocol and regulatory requirements Provide educational support
QUALITY ASSURANCE PROGRAM Elaine Armstrong, MS Quality Assurance Manager PURPOSE Verify accuracy of submitted data Verify compliance with protocol and regulatory requirements Provide educational support
Adverse Events: Thorough Analysis
 CMS TRANSPLANT PROGRAM QUALITY WEBINAR SERIES Adverse Events: Thorough Analysis James Ballard, MBA, CPHQ, CPPS, HACP Eileen Willey, MSN, BSN, RN, CPHQ, HACP QAPI Specialist/ Quality Surveyor Educators
CMS TRANSPLANT PROGRAM QUALITY WEBINAR SERIES Adverse Events: Thorough Analysis James Ballard, MBA, CPHQ, CPPS, HACP Eileen Willey, MSN, BSN, RN, CPHQ, HACP QAPI Specialist/ Quality Surveyor Educators
Toward Greater Scientific Rigor
 Toward Greater Scientific Rigor Good Clinical Practice (GCP) A NEW REQUIREMENT! The Center for Clinical & Translational Science 205.934.7442 ccts@uab.edu www.uab.edu/ccts @cctsnetwork The requirement:
Toward Greater Scientific Rigor Good Clinical Practice (GCP) A NEW REQUIREMENT! The Center for Clinical & Translational Science 205.934.7442 ccts@uab.edu www.uab.edu/ccts @cctsnetwork The requirement:
managing or activities.
 STANDARD OPERATING PROCEDURE Clinical Research Monitoring TITLE: Site Initiation Visit TITLE: Site Initiation Visit 1. PURPOSE SOP Number: Version: 1.0 MICHR CRM MON 002 Effective Date: 19Dec2013 1.1 This
STANDARD OPERATING PROCEDURE Clinical Research Monitoring TITLE: Site Initiation Visit TITLE: Site Initiation Visit 1. PURPOSE SOP Number: Version: 1.0 MICHR CRM MON 002 Effective Date: 19Dec2013 1.1 This
Signature Date Date First Effective: Signature Date Revision Date:
 University of Kentucky Office of Research Integrity and Institutional Review Board Standard Operating Procedures Revision #7 TITLE: NCI CIRB Review Page 1 of 15 C3.0400 Approved By: ORI Director Signature
University of Kentucky Office of Research Integrity and Institutional Review Board Standard Operating Procedures Revision #7 TITLE: NCI CIRB Review Page 1 of 15 C3.0400 Approved By: ORI Director Signature
Version Number: 004 Controlled Document Sponsor: Controlled Document Lead:
 Chief Investigators and Principal Investigators in Research Policy CONTROLLED DOCUMENT CATEGORY: CLASSIFICATION: PURPOSE Controlled Document Number: Policy Governance To set out the responsibilities of
Chief Investigators and Principal Investigators in Research Policy CONTROLLED DOCUMENT CATEGORY: CLASSIFICATION: PURPOSE Controlled Document Number: Policy Governance To set out the responsibilities of
Research Compliance Oversight in the Department of Veterans Affairs
 Research Compliance Oversight in the Department of Veterans Affairs Karen M. Smith, PhD Director, Midwestern Regional Office Office of Research Oversight Department of Veterans Affairs Health Care Compliance
Research Compliance Oversight in the Department of Veterans Affairs Karen M. Smith, PhD Director, Midwestern Regional Office Office of Research Oversight Department of Veterans Affairs Health Care Compliance
Inspections, Compliance, Enforcement, and Criminal Investigations
 1 of 5 2/11/2014 11:24 AM Home Inspections, Compliance, Enforcement, and Criminal Investigations Enforcement Actions Warning Letters Inspections, Compliance, Enforcement, and Criminal Investigations Baxter
1 of 5 2/11/2014 11:24 AM Home Inspections, Compliance, Enforcement, and Criminal Investigations Enforcement Actions Warning Letters Inspections, Compliance, Enforcement, and Criminal Investigations Baxter
Harrison Clinical Research. Monitoring of Clinical Trials - Quality Management from a CRO s Perspective
 Harrison Clinical Research Monitoring of Clinical Trials - Quality Management from a CRO s Perspective www.harrisonclinical.com 1 17Sep2009 Overview: Monitoring of Clinical Trials 1. Discovering and developing
Harrison Clinical Research Monitoring of Clinical Trials - Quality Management from a CRO s Perspective www.harrisonclinical.com 1 17Sep2009 Overview: Monitoring of Clinical Trials 1. Discovering and developing
USING SMART IRB AND SINGLE IRB REVIEW
 USING SMART IRB AND SINGLE IRB REVIEW Jeannie Barone Director, HRPO ATTRIBUTES Special thanks to Nichelle Cobb, PhD from University of Wisconsin-Madison for her permission to utilize her slides on SMART
USING SMART IRB AND SINGLE IRB REVIEW Jeannie Barone Director, HRPO ATTRIBUTES Special thanks to Nichelle Cobb, PhD from University of Wisconsin-Madison for her permission to utilize her slides on SMART
UConn Health Office of Clinical & Translational Research Standard Operating Procedures
 Purpose and Applicability: To ensure that a Medicare Coverage Analysis is done by staff in OCTR for all research clinical trials that produce r routine clinical services (RC) to be billed to Medicare and
Purpose and Applicability: To ensure that a Medicare Coverage Analysis is done by staff in OCTR for all research clinical trials that produce r routine clinical services (RC) to be billed to Medicare and
Medical Devices: Do I Need to Open a CAPA?
 Medical Devices: Do I Need to Open a CAPA? Adrienne Franco Busby, Partner, Faegre Baker Daniels LLP Anne Miller, Principal Regulatory Counsel, Medtronic Marta L. Villarraga, Principal, Exponent, Inc. Moderated
Medical Devices: Do I Need to Open a CAPA? Adrienne Franco Busby, Partner, Faegre Baker Daniels LLP Anne Miller, Principal Regulatory Counsel, Medtronic Marta L. Villarraga, Principal, Exponent, Inc. Moderated
11/18/2016. UC Irvine s Clinical Research Coordinator Certification Preparation Series PI Roles and Responsibilities SESSION 4
 UC Irvine s Clinical Research Coordinator Certification Preparation Series PI Roles and Responsibilities BEVERLY ALGER, CCRP, CHRC Research Compliance Officer Office of Research Compliance November 2016
UC Irvine s Clinical Research Coordinator Certification Preparation Series PI Roles and Responsibilities BEVERLY ALGER, CCRP, CHRC Research Compliance Officer Office of Research Compliance November 2016
Medicine Reconciliation FREQUENTLY ASKED QUESTIONS NATIONAL MEDICATION SAFETY PROGRAMME
 Medicine Reconciliation FREQUENTLY ASKED QUESTIONS NATIONAL MEDICATION SAFETY PROGRAMME The Process What is medicine reconciliation? Medicine reconciliation is an evidence-based process, which has been
Medicine Reconciliation FREQUENTLY ASKED QUESTIONS NATIONAL MEDICATION SAFETY PROGRAMME The Process What is medicine reconciliation? Medicine reconciliation is an evidence-based process, which has been
Successful FDA Inspections at Investigative Sites for Clinical Trials of Drugs and Biologics
 Vol. 9, No. 1, January 2013 Happy Trials to You Successful FDA Inspections at Investigative Sites for Clinical Trials of Drugs and Biologics By Swati Tendolkar The United States Food and Drug Administration
Vol. 9, No. 1, January 2013 Happy Trials to You Successful FDA Inspections at Investigative Sites for Clinical Trials of Drugs and Biologics By Swati Tendolkar The United States Food and Drug Administration
DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service 3%3&4
 DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service 3%3&4 JUL 1 3 2CG3 WARNING LETTER Food and Drug Administration Center for Devices and Radiological Health 2098 Gaither Road Rockville, MD 20850
DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service 3%3&4 JUL 1 3 2CG3 WARNING LETTER Food and Drug Administration Center for Devices and Radiological Health 2098 Gaither Road Rockville, MD 20850
EXEMPT RESEARCH. 1. Overview
 EXEMPT RESEARCH 1. Overview Research involving human subjects may be exempt from federal regulations requiring IRB review. The Ohio State University (HRPP) is responsible for determining whether research
EXEMPT RESEARCH 1. Overview Research involving human subjects may be exempt from federal regulations requiring IRB review. The Ohio State University (HRPP) is responsible for determining whether research
Marie-Claire Rickard, Governance and GCP Manager Jimena Lovos, Quality Assurance Manager Elizabeth Clough, R&D Governance Operations Manager
 Standard Operating Procedures (SOP) for: Reporting of Serious Breaches of or the Trial Protocol SOP Number: 037 Version Number: 5.0 Effective Date: 17/6/16 Review Date: 17/6/18 Author: Reviewer: Reviewer
Standard Operating Procedures (SOP) for: Reporting of Serious Breaches of or the Trial Protocol SOP Number: 037 Version Number: 5.0 Effective Date: 17/6/16 Review Date: 17/6/18 Author: Reviewer: Reviewer
IS YOUR QAPI COP READY?
 IS YOUR QAPI COP READY? Lisa Meadows/MSW Clinical Compliance Educator Accreditation Commission for Health Care OBJECTIVES Review the CMS requirements for the Medicare Condition of Participation: Quality
IS YOUR QAPI COP READY? Lisa Meadows/MSW Clinical Compliance Educator Accreditation Commission for Health Care OBJECTIVES Review the CMS requirements for the Medicare Condition of Participation: Quality
The SOP applies to all human subject research falling under the purview of the University of Missouri Institutional Review Board.
 Institutional Review Board.... University of Missouri-Columbia.. Standard Operating Procedure Informed Consent Types and Elements Informed Consent Types and Elements Effective Date: December 12, 2005 Original
Institutional Review Board.... University of Missouri-Columbia.. Standard Operating Procedure Informed Consent Types and Elements Informed Consent Types and Elements Effective Date: December 12, 2005 Original
Department of Defense Human Research Protection Program DOD INSTITUTIONAL AGREEMENT FOR INSTITUTIONAL REVIEW BOARD (IRB) REVIEW (IAIR)
 REVIEW (IAIR)") Department of Defense Human Research Protection Program DOD INSTITUTIONAL AGREEMENT FOR INSTITUTIONAL REVIEW BOARD (IRB) REVIEW (IAIR) General Instructions to Institutions and IRBs This form should be
Department of Defense Human Research Protection Program DOD INSTITUTIONAL AGREEMENT FOR INSTITUTIONAL REVIEW BOARD (IRB) REVIEW (IAIR) General Instructions to Institutions and IRBs This form should be
1. Department of Defense (DoD) Human Subjects Protection Regulatory Requirements
 Human Subjects Protection Regulatory Requirements") Information for Investigators: Headquarters, U.S. Special Operations Command Human Research Protection Office (HRPO) Human Research Protections Regulatory Requirements 1. Department of Defense (DoD) Human
Information for Investigators: Headquarters, U.S. Special Operations Command Human Research Protection Office (HRPO) Human Research Protections Regulatory Requirements 1. Department of Defense (DoD) Human